Static vs. Dynamic Headspace for Solvent Analysis: A Comprehensive Guide for Pharmaceutical Scientists
This article provides a detailed comparison of static and dynamic headspace gas chromatography techniques for the analysis of residual solvents in pharmaceuticals.
Static vs. Dynamic Headspace for Solvent Analysis: A Comprehensive Guide for Pharmaceutical Scientists
Abstract
This article provides a detailed comparison of static and dynamic headspace gas chromatography techniques for the analysis of residual solvents in pharmaceuticals. Tailored for researchers, scientists, and drug development professionals, it covers foundational principles, methodological applications, troubleshooting strategies, and a direct validation of both techniques against regulatory standards. Readers will gain a clear understanding of how to select, optimize, and apply the appropriate headspace method to ensure drug safety, comply with pharmacopeial methods like USP <467>, and improve analytical efficiency in quality control and R&D.
Understanding Headspace Fundamentals: Principles of Static and Dynamic Extraction
Headspace sampling is a premier sample preparation technique that leverages the volatility of certain compounds to isolate them from complex liquid or solid matrices for analysis by gas chromatography (GC). This guide provides an objective comparison of the two primary headspace techniques—static and dynamic—framed within solvent research for drug development. We synthesize current experimental data and methodologies to delineate the performance, advantages, and optimal application scenarios of each approach, providing scientists with a clear framework for selection and implementation.
Headspace (HS) sampling is a sample preparation technique designed to analyze the volatile fraction of a sample contained within a closed vial. The fundamental principle involves heating the sample to encourage volatile organic compounds (VOCs) to partition into the gas phase (the "headspace") above it [1] [2]. This vapor phase is then sampled and introduced into a gas chromatograph, effectively isolating volatile analytes from non-volatile matrix components such as proteins, salts, or polymers that could harm the analytical column or interfere with the analysis [1]. The technique is widely applicable across pharmaceuticals, environmental monitoring, food and beverage, and forensic science [3] [2].
The core advantage of headspace sampling lies in its simplicity and the cleanliness of the final extract. It eliminates extensive sample preparation, reduces solvent consumption (making it a "green" alternative), minimizes system maintenance, and extends the lifetime of the GC column [1] [2]. For drug development professionals, this translates to more robust and reproducible methods for analyzing residual solvents, reaction intermediates, and degradation products. This guide focuses on the comparison between the two main implementations of this principle: static and dynamic headspace sampling.
Static vs. Dynamic Headspace: A Technical Comparison
Static and dynamic headspace sampling differ fundamentally in their mechanics, which directly influences their sensitivity, application range, and operational complexity.
- Static Headspace (S-HS) involves heating a sample in a sealed vial until the volatile compounds reach equilibrium between the sample matrix and the headspace. Once equilibrium is established, a portion of the headspace vapor is withdrawn, typically via a gas-tight syringe or a valve-and-loop system, and injected into the GC [1] [3]. The process is characterized by a single extraction step from a closed system.
- Dynamic Headspace (D-HS), also known as Purge and Trap, continuously sweeps an inert gas over or through the sample, stripping volatile compounds from the matrix. The analytes are carried onto a trap containing an adsorbent material, where they are concentrated. After the collection period, the trap is heated to rapidly desorb the compounds into the GC [4] [2]. This is a multi-step, non-equilibrium technique designed for exhaustive extraction.
The table below summarizes the core differences between the two techniques.
Table 1: Fundamental Characteristics of Static and Dynamic Headspace
| Feature | Static Headspace (S-HS) | Dynamic Headspace (D-HS) |
|---|---|---|
| Basic Principle | Equilibrium-based single extraction from a closed system [3] | Continuous extraction (non-equilibrium) and concentration onto a trap [4] |
| Typical Setup | Valve-and-loop or heated syringe [1] [2] | Multi-bed sorbent trap [2] |
| Key Advantage | Simplicity, robustness, high reproducibility [1] | Superior sensitivity and trace-level detection [2] |
| Sample Throughput | Generally high and easily automated | Can be lower due to longer trap desorption times |
| Solvent Consumption | Minimal to none (green technique) [3] | None for extraction, may require solvent for trap desorption in some setups [5] |
Performance Comparison: Experimental Data and Applications
The theoretical differences between S-HS and D-HS manifest distinctly in their analytical performance. Selecting the appropriate technique depends on the specific requirements of the analysis, particularly regarding sensitivity and the nature of the target analytes.
Sensitivity and Analytical Scope
Dynamic headspace generally provides significantly higher sensitivity than static headspace. This is because D-HS continuously removes and concentrates analytes from the sample, effectively enriching trace components that would yield a signal below the detection limit in a single S-HS extraction [2]. A comparative study of an ancient perfume resin found that Solid-Phase Microextraction (SPME), a static technique, provided a more extensive dataset of olfactory compounds compared to standard S-HS, attributed to the concentration effect of the fiber [6]. However, for true trace-level analysis, full D-HS is often superior.
Furthermore, D-HS is suited to a wider range of volatile and semi-volatile compounds due to the availability of various adsorbent traps [2]. Static systems, in contrast, are highly sensitive for low-boiling point compounds but may struggle with heavier semi-volatiles [2].
Quantitative Reproducibility and Greenness
Static headspace excels in reproducibility for quantitative analysis. Its equilibrium-based nature, when parameters are well-controlled, ensures highly consistent results, which is why it is the preferred method for standardized tests like residual solvent analysis in pharmaceuticals (e.g., USP <467>) [1]. A 2025 study on volatile petroleum hydrocarbons (VPHs) demonstrated that a statistically optimized S-HS method could achieve excellent reproducibility, confirming its fitness for regulatory environmental monitoring [7].
From a green chemistry perspective, both techniques are advantageous as they drastically reduce or eliminate solvent use compared to liquid-liquid extraction [3] [8]. A greenness evaluation using the AGREEprep tool confirmed the strong alignment of SPME-based methods with green principles, highlighting benefits like solvent elimination, low waste, and high operator safety [8].
Table 2: Performance Comparison and Typical Applications
| Aspect | Static Headspace (S-HS) | Dynamic Headspace (D-HS) |
|---|---|---|
| Sensitivity | Good for moderate-concentration VOCs [7] | Excellent for trace-level and ultra-trace analysis [2] |
| Reproducibility | Excellent (equilibrium-driven) [7] [1] | Good, though can be influenced by trap stability and carryover |
| Application Examples | Residual solvents in APIs [1], blood alcohol [1], VOCs in water [7] | Odor profiling in food [3], ignitable liquid residues in fire debris [5], trace VOCs in plastics/rubber [3] |
| Greenness | High (solvent-free, minimal waste) [3] | High (solvent-free); can eliminate neurotoxic solvents like carbon disulfide [5] |
Experimental Protocols for Method Optimization
Robust headspace analysis requires careful optimization of key parameters. The following protocols, drawn from recent studies, provide a blueprint for developing reliable methods.
Protocol 1: Multivariate Optimization of Static Headspace for Aqueous Matrices
A 2025 study optimized a static HS-GC-FID method for C5–C10 volatile petroleum hydrocarbons in water using a Central Composite Face-centered (CCF) experimental design [7]. This approach efficiently models interaction effects between parameters, which traditional one-variable-at-a-time (OVAT) methods miss.
- Instrumentation: Agilent 6890 GC-FID system coupled with a static headspace sampler (Agilent G1888), using a DB-1 capillary column. Helium was the carrier gas [7].
- Sample Preparation: 20 mL headspace vials were used. Samples were prepared with ultrapure water, spiked with hydrocarbon standards, and supplemented with 1.8 g of NaCl to improve partitioning into the headspace via salting-out. The methanol concentration was kept below 1% v/v to avoid affecting the equilibrium [7].
- Parameters Optimized: The experimental design simultaneously varied sample volume, equilibration temperature, and equilibration time. The response variable was the chromatographic peak area per microgram of analyte [7].
- Key Findings: Analysis of variance (ANOVA) confirmed the model's global significance (R² = 88.86%, p < 0.0001). Sample volume showed the strongest negative impact, while temperature and its interactions with time demonstrated synergistic behavior. The resulting optimized method was validated and shown to be sensitive and reproducible for environmental monitoring [7].
Protocol 2: Dynamic Headspace for Complex Solid Matrices
Dynamic headspace is particularly valuable for complex solid matrices where exhaustive extraction is needed. A National Institute of Justice report detailed the use of Dynamic Vapor Microextraction (DVME), a small-volume purge and trap method, for extracting ignitable liquid residues from fire debris [5].
- Instrumentation: The DVME system uses a short (1-3 m) section of porous layer open tubular (PLOT) capillary as a trap, coated with an adsorbent material [5].
- Sample Preparation: Fire debris is placed in a sealed container. The DVME system pulls the headspace vapor through the PLOT capillary, where volatile compounds are concentrated.
- Extraction & Desorption: The dynamic extraction runs for a set time or volume. Subsequently, the trap is heated, and the analytes are desorbed using a small volume of a relatively benign solvent like acetone. This contrasts with the traditional activated carbon strip method, which requires toxic carbon disulfide for elution [5].
- Key Advantage: This protocol highlights how D-HS can provide a cleaner, safer, and non-invasive alternative to older methods while still achieving the high sensitivity required for forensic analysis.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful headspace analysis relies on several key consumables and materials. The following table details essential items for a typical headspace experiment.
Table 3: Essential Research Reagents and Materials for Headspace Analysis
| Item | Function | Examples & Notes |
|---|---|---|
| Headspace Vials | Sealed container to hold sample and maintain equilibrium [3] | 20 mL vials are common; must be sealed with PTFE/silicone septa and aluminum crimp caps to prevent analyte loss [7]. |
| Salting-Out Agents | Modifies ionic strength to improve VOC partitioning from aqueous phases [3] | Sodium Chloride (NaCl); reduces solubility of hydrophobic VOCs, increasing their concentration in the headspace [7]. |
| Internal Standards | Corrects for analytical variability and quantitation errors. | Isotope-labeled analogs of target analytes are ideal for mass spectrometry. |
| Sorbent Materials | For trapping and concentrating volatiles in D-HS and SPME [3] [8]. | Tenax TA, Carbopack, activated carbon; for SPME, common coatings include PDMS, DVB/PDMS, CAR/PDMS [3]. |
| Adjusted Solvents | Dissolves samples without interfering; used when sample is not directly amenable. | Water, DMSO, DMF; must be low-volatility to avoid overwhelming the detector [1]. |
The choice between static and dynamic headspace sampling is not a matter of one being universally superior, but rather of selecting the right tool for the specific analytical challenge. Static headspace is the method of choice for robust, reproducible, and high-throughput quantitative analysis of relatively abundant volatile compounds, as seen in pharmaceutical quality control [1]. Dynamic headspace is indispensable for pushing the boundaries of sensitivity, enabling the detection and identification of trace-level volatiles and semi-volatiles in complex matrices like polymers, biological fluids, and fire debris [3] [5].
Future trends point towards increased automation, the development of novel sorbent materials for broader selectivity, and the deeper integration of multivariate statistical designs for method optimization [7] [9]. Furthermore, the coupling of non-separative headspace to mass spectrometry (HS-MS) for rapid sample fingerprinting and the refinement of green, solvent-free techniques like TWA-SPME will continue to expand the utility of headspace sampling in drug development and beyond [4] [8]. For the modern laboratory, a thorough understanding of both static and dynamic principles is essential for constructing a versatile and effective analytical toolkit.
Visual Appendix
Static Headspace Workflow
Dynamic Headspace Workflow
Fundamental Principles and Mathematical Foundation
Static Headspace Extraction (SHE) is a widely used sample introduction technique for gas chromatography (GC) that relies on the fundamental principle of thermodynamic equilibrium between a sample and the vapor phase above it in a sealed vial [10] [11]. The technique is prized for its simplicity, minimal sample preparation, and ability to analyze volatile compounds in complex matrices without introducing non-volatile contaminants into the GC system [12] [11].
At the heart of static headspace analysis lies the partition coefficient (K), which quantitatively describes the distribution of an analyte between the sample phase and the gas phase at equilibrium [10] [11]. This coefficient is defined as the ratio of the analyte's concentration in the sample phase (CS) to its concentration in the gas phase (CG): K = CS/CG [11]. The partition coefficient is a temperature-dependent parameter that reflects the affinity of an analyte for the sample matrix compared to the vapor phase. Analytes with low K values have a higher tendency to partition into the headspace, making them more easily detectable, while those with high K values remain primarily in the sample matrix, presenting an analytical challenge [10].
The relationship between the initial analyte concentration in the sample and the final detector response is mathematically described by the fundamental static headspace equation [10] [11]:
A ∝ CG = C0/(K + β)
Where:
- A is the chromatographic peak area (detector response)
- CG is the concentration of analyte in the gas phase
- C0 is the initial concentration of analyte in the sample
- K is the partition coefficient
- β is the phase ratio (volume of gas phase/volume of sample phase)
This equation reveals that to maximize detector response (A), the sum of K and β in the denominator must be minimized [11]. The phase ratio (β) represents the relative volumes of the headspace and sample within the sealed vial, typically ranging between 1-20 in most SHE methods [10]. When the partition coefficient and phase ratio are of similar magnitude, the phase ratio significantly impacts the final peak area, whereas when K is much larger than β, the phase ratio has negligible effect [10].
Figure 1: The Static Headspace Equilibrium Process. This diagram illustrates the establishment of equilibrium between the sample and headspace phases in a sealed vial, and how this relationship defines the partition coefficient (K), which in turn determines the analyte concentration in the gas phase (CG) and ultimately the chromatographic detector response (A).
Experimental Parameters and Method Optimization
Successful implementation of static headspace analysis requires careful optimization of several key parameters that influence the partition coefficient and the efficiency of analyte transfer to the gas phase.
Temperature Optimization
Temperature profoundly affects static headspace analysis by directly influencing the partition coefficient [10] [11]. Increasing the vial temperature reduces the partition coefficient (K) for most analytes, shifting the equilibrium toward the vapor phase and resulting in higher analyte concentrations in the headspace [10]. This relationship follows fundamental thermodynamic principles where increased thermal energy promotes vaporization of analytes. However, temperature optimization requires balancing sensitivity gains against potential risks, including solvent vaporization, analyte degradation, or changes in matrix effects [10] [11]. As a general guideline, the maximum oven temperature should be maintained approximately 20°C below the solvent's boiling point to prevent excessive pressure buildup and solvent vaporization [11].
The impact of temperature on the partition coefficient can be substantial. For example, the K value for ethanol in water decreases from approximately 1350 at 40°C to about 330 at 80°C, significantly enhancing detector response at higher temperatures [11]. Strong solute-solvent interactions or matrix effects can attenuate the influence of temperature on vaporization, particularly for polar analytes in polar matrices [10] [13]. In some specific cases, such as non-polar solutes dissolved in polar solvents at low concentrations, matrix effects can actually enhance vaporization as the non-polar solute is repelled by the polar solvent [10].
Phase Ratio and Sample Volume
The phase ratio (β), defined as the ratio of headspace volume to sample volume (VG/VS), significantly impacts method sensitivity, particularly for analytes with low partition coefficients [10] [11]. A best practice for optimizing phase ratio is to maintain at least 50% headspace in the vial [11]. Reducing the phase ratio (by increasing sample volume or using smaller vials) generally enhances sensitivity for volatile analytes by increasing the proportion of analyte in the headspace at equilibrium [10] [11].
The effect of phase ratio on sensitivity depends on the relationship between K and β [10]:
- When K ≈ β, both the partition coefficient and phase ratio significantly influence peak area
- When K >> β, the phase ratio has minimal effect on final peak area
- When K << β, the phase ratio dramatically affects peak area, requiring careful sample volume control
Experimental data demonstrates that using a 20-mL vial instead of a 10-mL vial with the same 4-mL sample volume improves detector response due to the more favorable phase ratio [11]. Similarly, increasing sample volume within the same vial size decreases β and enhances sensitivity for volatile analytes [11]. However, excessively small sample volumes with volatile solvents risk complete evaporation during heating, compromising reproducibility [10].
Equilibrium Time and Matrix Modification
Equilibrium time must be experimentally determined for each sample type, as insufficient equilibration is a leading cause of reproducibility issues in SHE [10] [11]. The time required to establish equilibrium varies significantly based on sample matrix, analyte properties, temperature, and agitation conditions. Modern automated headspace instruments often include tools to help determine optimal equilibration times experimentally [11].
Matrix modification techniques can enhance method sensitivity by altering the partition coefficient:
- Salting-out: Adding non-volatile salts like ammonium sulfate or sodium chloride to aqueous samples reduces analyte solubility, pushing more analyte into the headspace [13] [14]
- Co-solvent addition: Incorporating small amounts of co-solvents or higher boiling point diluents can modify solvent polarity and promote analyte partitioning into the headspace, particularly for non-polar matrices [14]
- pH adjustment: For ionizable compounds, pH modification can suppress ionization and enhance volatility
Agitation intensity during incubation also promotes mass transfer between phases by disrupting boundary layers, potentially reducing the time required to reach equilibrium [13].
Comparison with Dynamic Headspace Extraction
While both static and dynamic headspace extraction aim to isolate volatile compounds for GC analysis, they employ fundamentally different principles and offer distinct advantages and limitations.
Table 1: Systematic Comparison of Static vs. Dynamic Headspace Extraction
| Feature | Static Headspace Extraction | Dynamic Headspace Extraction |
|---|---|---|
| Fundamental Principle | Equilibrium-based sampling [10] [12] | Continuous purging with inert gas [12] [15] |
| Sensitivity | Good for volatiles at high ppb levels [10] [16] | 50-100x higher sensitivity; suitable for trace-level analysis [15] [16] |
| Extraction Yield | Typically 10-20% [16] | Up to 80% [16] |
| Method Detection Limits | ~100 ng/L [16] | Picogram per liter range [16] |
| Typical Applications | Residual solvents, flavors, VOCs in environmental samples [12] [11] | Trace analysis in water, air, solids; complex matrices [12] [13] |
| Reproducibility | RSDs <27% [16] | RSDs <27% [16] |
| Matrix Compatibility | Limited for solid, polar, or complex matrices [13] [14] | Excellent for solid samples and complex matrices [12] [14] |
| Equipment Complexity | Simple [12] [2] | More complex, requires traps and gas flow systems [12] [14] |
Dynamic Headspace Extraction (DHE), also known as purge and trap, does not rely on equilibrium establishment but instead uses continuous purging with an inert gas to sweep volatiles from the sample onto a sorbent trap [10] [12]. This approach provides significantly enhanced sensitivity through analyte preconcentration, with extraction yields reaching 80% compared to 10-20% for static techniques [16]. The continuous removal of volatiles according to Le Chatelier's Principle enables DHE to qualitatively extract nearly all analyte from a sample, making it particularly valuable for ultratrace analysis [10].
Advanced variants of dynamic headspace have been developed to address specific analytical challenges:
- Full Evaporative Technique (FET): The sample and matrix are completely evaporated before collection onto an adsorbent trap, particularly useful for volatile compounds in difficult-to-analyze matrices [13] [14]
- Multi Volatile Method (MVM): Employs sequential dynamic headspace extractions under different conditions to comprehensively profile analytes with diverse chemistries [13] [14]
Table 2: Quantitative Performance Comparison of Headspace Techniques
| Technique Category | Specific Technique | Extraction Yield | Method Detection Limits | Relative Standard Deviation |
|---|---|---|---|---|
| Static Sampling | Syringe or loop | ~10-20% [16] | ~100 ng/L [16] | <27% [16] |
| Static Enrichment | SPME, PAL SPME Arrow | Up to 80% [16] | Picogram/L range [16] | <27% [16] |
| Dynamic Enrichment | ITEX, Trap sampling | Up to 80% [16] | Picogram/L range [16] | <27% [16] |
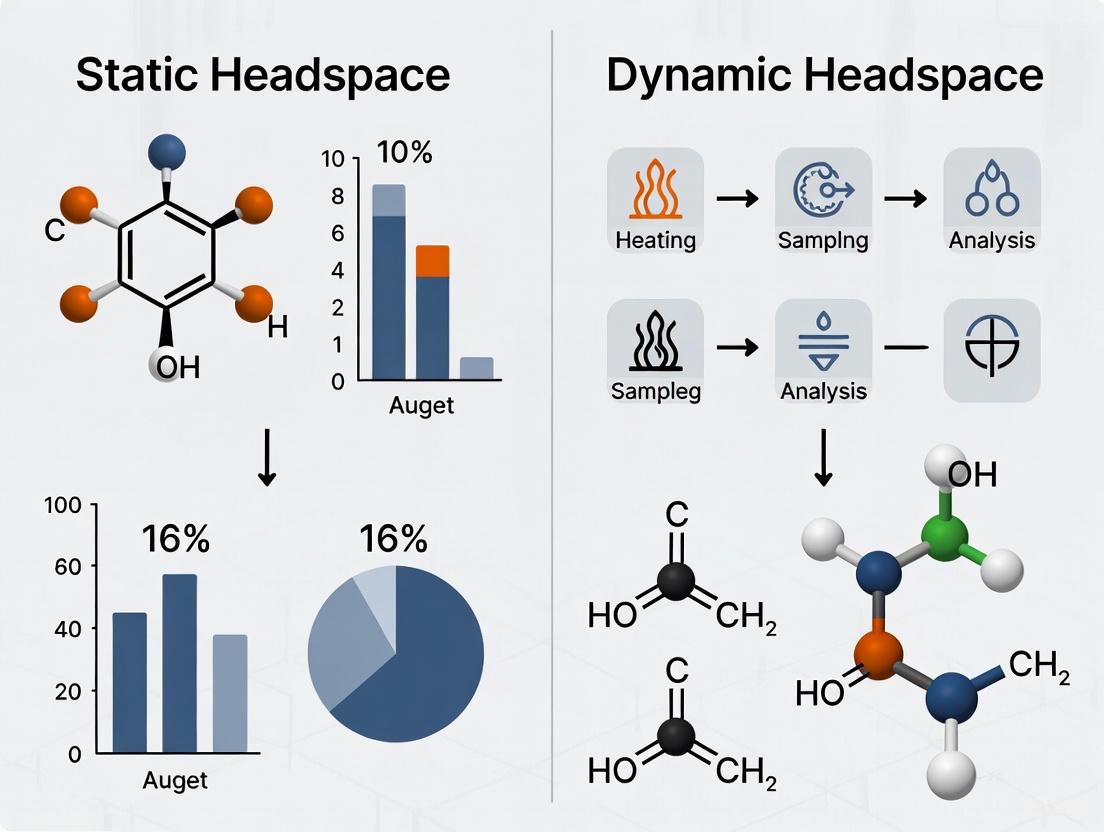
Figure 2: Static vs. Dynamic Headspace Experimental Workflows. This diagram compares the fundamental procedural differences between static and dynamic headspace techniques, highlighting their distinct operational steps and optimal application scenarios.
Essential Research Reagent Solutions
Successful implementation of static headspace analysis requires specific materials and reagents optimized for volatile compound analysis.
Table 3: Essential Research Reagents and Materials for Static Headspace Analysis
| Item | Function | Application Notes |
|---|---|---|
| Headspace Vials | Contain sample and maintain sealed environment during equilibration [11] | Common sizes: 10-mL, 20-mL, 22-mL; must provide reliable seal [11] |
| Septum & Caps | Create airtight seal to prevent volatile loss [11] | Crimp or screw tops; critical for maintaining equilibrium [10] [11] |
| Salting-Out Agents | Reduce analyte solubility in aqueous matrices [13] [14] | Ammonium sulfate, sodium chloride; enhance volatile partitioning [14] |
| Co-solvents/Additives | Modify matrix polarity to enhance volatile release [14] | Higher boiling point diluents; promote analyte partitioning [14] |
| Calibration Standards | Quantitative method development and validation | Should match matrix composition when possible [11] |
| Internal Standards | Correct for analytical variability | Deuterated analogs or similar compounds not present in sample |
Application Protocols and Experimental Design
Standard Static Headspace Protocol for Residual Solvents
This protocol outlines a generalized methodology for analyzing residual solvents in pharmaceutical products using static headspace GC, adaptable to various sample types with appropriate modifications.
Sample Preparation: Precisely weigh approximately 100-500 mg of sample into a 20-mL headspace vial. For solid samples, consider particle size reduction to increase surface area. Add appropriate internal standard if required for quantification.
Matrix Modification: If analyzing aqueous samples, add salting-out agent (e.g., 1-3 g ammonium sulfate) to enhance volatile partitioning [14]. For non-polar matrices, consider adding small amounts of co-solvents to modify polarity.
Vial Sealing: Immediately seal the vial with appropriate septum and cap to prevent volatile loss. Ensure tight seal to maintain system integrity during heating.
Equilibration Conditions: Place vials in automated headspace sampler or temperature-controlled block. Typical conditions: 70-90°C for 15-45 minutes with agitation [11] [13]. Specific temperatures should remain 20°C below solvent boiling point [11].
Pressure Equilibrium: Pressurize vial with carrier gas to facilitate transfer to GC inlet. Typical pressurization times: 0.5-2 minutes depending on system.
Sample Transfer: Open transfer line to GC inlet for precisely controlled time (typically 0.1-1.0 minutes) to inject headspace aliquot.
Chromatographic Conditions: Use appropriate GC column (e.g., mid-polarity stationary phase) and optimized temperature program for target analytes. Detection typically via FID or MS.
Quantitation: Employ external standard calibration or internal standard method for highest accuracy. For challenging matrices, consider Multiple Headspace Extraction (MHE) to address matrix effects [11].
Method Development Considerations
Effective static headspace method development requires systematic optimization of key parameters:
Equilibration Time Determination: Conduct time-profile experiments by analyzing replicates at different equilibration times (e.g., 10, 20, 30, 45, 60 minutes) while holding other parameters constant. The optimal time is when analyte responses plateau, indicating equilibrium establishment [11].
Temperature Optimization: Perform temperature ramping studies (e.g., 50, 60, 70, 80, 90°C) to identify the temperature that maximizes response without causing solvent vaporization or analyte degradation [11].
Sample Volume Optimization: Test different sample volumes (e.g., 1, 2, 3, 5 mL in 20-mL vial) to determine the phase ratio that provides optimal sensitivity while maintaining at least 50% headspace [11].
Salting-Out Efficiency: When analyzing aqueous samples, compare different salts (ammonium sulfate vs. sodium chloride) and concentrations to maximize volatile partitioning into headspace [14].
For complex methods with multiple interacting variables, experimental design (DoE) approaches such as factorial designs or response surface methodologies are recommended to efficiently identify optimal conditions and interactions [13] [14].
Static Headspace Extraction remains a powerful and widely applicable technique for volatile compound analysis, with its fundamental operation governed by the equilibrium-based partitioning of analytes between sample and vapor phases as described by the partition coefficient (K). While SHE offers simplicity, minimal sample preparation, and robust performance for many applications, its limitations in sensitivity and matrix compatibility necessitate alternative approaches like Dynamic Headspace Extraction for trace-level analysis or complex matrices. Understanding the core principle of equilibrium establishment and the mathematical relationship between the partition coefficient, phase ratio, and detector response enables researchers to effectively develop, optimize, and troubleshoot static headspace methods across diverse application areas including pharmaceutical analysis, environmental monitoring, and food and flavor characterization. The choice between static and dynamic approaches should be guided by analytical requirements, with static methods excelling in routine analysis of relatively concentrated volatile analytes, and dynamic techniques providing the sensitivity and comprehensive extraction needed for challenging applications at trace concentrations.
Headspace (HS) sampling is a premier technique for isolating volatile organic compounds (VOCs) from solid or liquid samples for gas chromatographic analysis, valued for being a green, solvent-free approach [4]. This methodology is fundamentally divided into two operational modes: static headspace and dynamic headspace. Static headspace relies on establishing equilibrium in a closed vial, where an aliquot of the vapor phase is sampled for analysis [10]. In contrast, dynamic headspace—also widely known as Purge and Trap—employs a continuous flow of inert gas to purge VOCs from the sample, followed by their concentration on an adsorbent trap before transfer to the gas chromatograph [17] [18]. The core principle of dynamic headspace is this continuous process of purging, trapping, and analyte transfer, which enables exhaustive extraction and superior sensitivity for trace-level volatile compounds [17] [19]. This guide provides an objective comparison of these techniques, focusing on their core principles, performance data, and applications relevant to researchers and scientists in solvents research and drug development.
Core Principles and Instrumentation
The Mechanism of Static Headspace
Static Headspace is an equilibrium-based technique. A sample is placed in a sealed vial and heated to a specific temperature until the volatile analytes distribute between the sample matrix and the vapor phase (headspace) above it [10]. After equilibrium is established, a portion of this headspace vapor is injected into the GC system, typically using a gas-tight syringe or a pressurized transfer line [10]. The theoretical foundation is described by the partition coefficient (K), which expresses the ratio of the analyte's concentration in the sample phase to its concentration in the gas phase at equilibrium [10]. The resulting peak area in the chromatogram is proportional to the initial analyte concentration, the partition coefficient, and the phase ratio (the volume of vapor phase divided by the volume of sample phase) [10]. This method is characterized by its simplicity, robustness, and minimal sample preparation.
The Mechanism of Dynamic Headspace
Dynamic Headspace is a non-equilibrium, exhaustive extraction technique. Its core principle involves three sequential and continuous operations, as illustrated in the workflow below:
- Purging: An inert gas, such as helium or nitrogen, is continuously bubbled through the liquid sample or passed over a solid sample. This action strips volatile compounds from the sample matrix by constantly removing them from the headspace, preventing equilibrium from being established. According to Le Chatelier's Principle, this encourages continuous migration of volatiles from the liquid to the vapor phase, resulting in a more complete extraction than static methods [17] [10]. The efficiency of this purge is governed by factors like sweep volume, which is a function of purge time and gas flow rate [17].
- Trapping: The purge gas, now laden with VOCs, is directed through an adsorbent trap. This trap is essentially a short GC column packed with materials that retain the organic compounds. Water vapor, which is often co-purged, is typically managed to minimize interference [17]. The trap is often designed with multiple adsorbents of varying strengths layered to effectively capture a wide range of volatilities; weaker adsorbents on top retain less volatile analytes, while stronger adsorbents below capture the more volatile ones, ensuring nothing is lost [17].
- Desorption and Transfer: After a predetermined purge time, the trap is rapidly heated while being backflushed with carrier gas. This thermal desorption releases the concentrated analytes and transfers them as a narrow, focused band into the gas chromatograph for separation and detection [17] [18]. This step is critical for maintaining chromatographic resolution.
Performance Comparison: Experimental Data
A systematic comparison of automated headspace techniques for GC analysis of VOCs in water provides robust quantitative data, summarized in the table below [16]. The techniques are classified into three categories: static sampling, static enrichment, and dynamic enrichment, which includes Dynamic Headspace/Purge and Trap.
Table 1: Performance Comparison of Headspace Sampling Techniques for VOC Analysis in Water [16]
| Technique Class | Specific Technique | Typical Extraction Yield (%) | Typical Method Detection Limit (MDL) | Relative Standard Deviation (RSD) |
|---|---|---|---|---|
| Static Sampling | Syringe / Loop | ~10 - 20% | ~100 ng/L | < 27% |
| Static Enrichment | SPME / SPME Arrow | Up to ~80% | Picogram/L (pg/L) range | < 27% |
| Dynamic Enrichment | ITEX / Trap (Purge & Trap) | Up to ~80% | Picogram/L (pg/L) range | < 27% |
This data demonstrates that dynamic enrichment techniques achieve significantly higher extraction yields and lower detection limits compared to static sampling. The exhaustive nature of dynamic headspace allows for up to 80% of the analytes to be extracted, enabling detection at trace levels [16].
Further evidence from a study analyzing nitrous oxide in seawater confirms this performance advantage. The study, which utilized a TCT/PTI Chrompack CP4010 sampling apparatus, found that Purge and Trap (PT) offered superior performance over Dynamic Headspace (DH) for this specific application [19].
Table 2: Comparison of Dynamic Headspace vs. Purge and Trap for Nitrous Oxide in Seawater [19]
| Technique | Extraction Recovery | Sensitivity | Detection Limit | Key Application |
|---|---|---|---|---|
| Dynamic Headspace (DH) | Lower than PT | Lower than PT | Higher than PT | Trace analysis in marine samples |
| Purge and Trap (PT) | Better | Better | Very low pmol/mL levels | Trace analysis in marine samples |
The study concluded that Purge and Trap combined with GC–ECD provided better extraction recovery, sensitivity, and detection limits, making it the more suitable methodology for ultratrace analysis [19].
Comparative Advantages and Application Suitability
The choice between static and dynamic headspace is driven by analytical requirements. The following diagram illustrates the decision-making logic for technique selection:
Beyond the core principle of sensitivity, the two techniques exhibit distinct operational profiles:
Table 3: Advantages and Disadvantages of Static vs. Dynamic Headspace
| Aspect | Static Headspace | Dynamic Headspace / Purge & Trap |
|---|---|---|
| Principle | Equilibrium | Exhaustive |
| Sensitivity | Moderate (~100 ng/L) [16] | High (picogram/L) [16] |
| Matrix Flexibility | Good for simple liquids | Excellent for solids, liquids, viscous samples [18] |
| Water Management | Minimal issue | Requires careful control to avoid trap damage/icing [17] [18] |
| Maintenance | Low | Higher (trap lifespan can be a factor) [18] |
| Quantitation | Straightforward, relies on equilibrium | Excellent, but requires careful calibration |
| Best For | Relatively clean samples with high VOC concentrations, routine analysis [10] | Complex matrices, ultratrace analysis, targeted low-level compounds [19] [18] |
As indicated in the diagram and table, Static Headspace is ideal for routine applications where analyte concentrations are relatively high, such as determining residual solvents in pharmaceuticals or ethanol in blood [10]. Its simplicity and lower maintenance make it suitable for high-throughput labs.
Conversely, Dynamic Headspace is the superior choice for demanding applications such as environmental monitoring of trace VOCs in water, flavor and fragrance analysis where detecting subtle compounds is crucial, and forensic science where sample matrices can be highly variable and complex [19] [18]. Its flexibility with sample matrices and ability to perform exhaustive extraction give it a distinct advantage in these areas, despite its more complex instrumentation.
Essential Research Reagent Solutions
The following table details key materials and reagents essential for conducting dynamic headspace analysis, based on the protocols and technical descriptions from the search results.
Table 4: Key Research Reagent Solutions for Dynamic Headspace
| Item | Function / Specification | Application Notes |
|---|---|---|
| Purge Gas | High-purity Inert Gas (Helium, Nitrogen) | Transports VOCs from sample to trap; inertness prevents oxidation [17] [18]. |
| Adsorbent Trap | Multi-bed sorbent (e.g., Tenax, Carbopack, Silica Gel) | Concentrates and focuses VOCs; layered beds handle a wide volatility range [17] [4]. |
| Internal Standards | Deuterated or stable isotope-labeled VOCs | Corrects for analytical variability and quantifies recovery [19]. |
| Salting-Out Agents | Saturated salt solutions (NaCl, KCl, NaNO₃) | Increases ionic strength, reduces VOC solubility, improves purge efficiency [20]. |
| Standard Solutions | Certified VOC mixtures in appropriate solvents | Used for system calibration, quantification, and method validation [20]. |
Within the context of solvent research and drug development, the choice between static and dynamic headspace is not a matter of one being universally better, but of selecting the right tool for the specific analytical challenge. Static Headspace offers simplicity, speed, and robustness for analyzing samples with higher concentrations of volatile compounds, making it a mainstay for quality control, such as residual solvent testing in active pharmaceutical ingredients. In contrast, Dynamic Headspace, with its core principle of continuous purging, trapping, and analyte transfer, provides an exhaustive extraction that delivers exceptional sensitivity and precision for ultratrace analysis. It is indispensable for identifying and quantifying low-level volatile impurities, degradation products, or trace contaminants in complex matrices. Understanding these fundamental principles and performance characteristics enables scientists to make an informed decision that aligns with their research objectives, ensuring data quality and analytical efficiency.
Headspace Gas Chromatography (HS-GC) is an indispensable technique for analyzing volatile organic compounds, particularly residual solvents, in pharmaceutical products. Its primary advantage lies in the introduction of a clean, vaporized sample into the GC, thereby preventing non-volatile matrix components from contaminating the inlet and column [21]. For researchers and drug development professionals, the reliability of quantitative results depends on a mastery of the closed chemical system within the headspace vial. Two parameters are paramount in controlling the sensitivity of this analysis: the equilibration temperature and the phase ratio (β). This guide objectively examines the governing principles of these parameters in static headspace systems and contrasts them with the alternative approach of dynamic headspace sampling, providing structured experimental data to inform method development.
Theoretical Foundations of Static Headspace Analysis
The Headspace Equilibrium System
In static headspace analysis, a sealed vial contains the sample (liquid or solid) and a headspace gas phase. Upon heating, volatile analytes partition between the sample matrix and the gas phase until equilibrium is established [22]. The fundamental relationship describing this system is defined by the following equation, which dictates the concentration of an analyte in the gas phase ((C_G)) that is ultimately transferred to the GC for detection:
(CG = C0 / (K + \beta)) [22] [21] [23]
Where:
- (C_G) = Analyte concentration in the gas phase (headspace)
- (C_0) = Original analyte concentration in the sample solution
- (K) = Partition coefficient ( (CS / CG) )
- (\beta) = Phase ratio ( (VG / VS) )
- (C_S) = Analyte concentration in the sample liquid phase
- (V_G) = Volume of the gas phase (headspace)
- (V_S) = Volume of the sample or liquid phase
The detector response is directly proportional to (C_G). Therefore, to maximize sensitivity, the sum (K + \beta) must be minimized [22] [21]. The following diagram illustrates this closed chemical system and the key parameters.
Core Parameters Governing Sensitivity
The partition coefficient ((K)) and phase ratio ((\beta)) are the two pillars of headspace sensitivity.
- Partition Coefficient (K): This compound- and matrix-specific constant describes the distribution of an analyte between the liquid and gas phases at equilibrium [21]. A high (K) value indicates the analyte favors the liquid phase (e.g., ethanol in water), resulting in a lower headspace concentration. A low (K) value indicates the analyte favors the gas phase (e.g., n-hexane in water), leading to a higher headspace concentration [24] [21].
- Phase Ratio (β): This is a physical parameter defined as the ratio of the headspace volume ((VG)) to the sample volume ((VS)) [21]. It is easily manipulated by the analyst by changing the sample volume or the vial size [22].
The relative magnitudes of (K) and (\beta) determine how temperature and sample volume changes will affect the sensitivity for different types of analytes [21].
Experimental Investigation of Key Parameters
A rigorous understanding of temperature and phase ratio is best achieved through controlled experiments. The following protocols and data illustrate their effects.
Protocol 1: Investigating the Temperature-Partition Coefficient Relationship
Objective: To determine the optimal equilibration temperature for the analysis of a soluble (ethanol) and a sparingly soluble (n-hexane) analyte in water.
Materials & Reagents:
- Standard Solutions: Aqueous solutions of ethanol and n-hexane at known concentrations.
- Headspace Vials: 20 mL vials with PTFE/silicone septa and aluminum crimp caps.
- Diluent: High-purity water.
- Internal Standard: (Optional) n-propanol for quantification [25].
Method:
- Sample Preparation: Pipette 10 mL of each standard solution into separate 20 mL headspace vials ((\beta = 1)). Seal vials immediately [26].
- HS-GC Conditions:
- Analysis: For each temperature, analyze six replicates ((n=6)). Record the peak areas for ethanol and n-hexane.
Results and Interpretation: The experimental data will reveal a stark contrast between the two analytes, as summarized in the table below.
Table 1: Experimental Impact of Temperature on Analyte Response
| Analyte | Partition Coefficient (K) | Impact of Temperature Increase on K | Impact on GC Peak Area | Theoretical Basis |
|---|---|---|---|---|
| Ethanol | High (~1350 at 40°C; ~330 at 80°C) [22] | Strong decrease [22] | Large increase (e.g., 6.3x from 40°C to 80°C) [21] | K >> β. Lowering K dominates, significantly increasing (C_G) [21]. |
| n-Hexane | Low (~0.01) [24] | Minimal change [21] | Small increase [21] | K << β. Change in K has little effect on the sum (K + β) [21]. |
As temperature increases, (K) decreases for most analytes, driving more analyte into the headspace. However, this effect is most dramatic for soluble analytes with high initial (K) values [24] [21]. The temperature must be controlled to within ±0.1°C for high-K analytes to achieve a precision of 5% [24]. The maximum temperature is typically set to 20°C below the boiling point of the solvent or sample matrix to avoid excessive pressure [22] [26].
Protocol 2: Investigating the Effect of Phase Ratio (β)
Objective: To evaluate the effect of changing sample volume (and thus (\beta)) on the headspace concentration of ethanol and n-hexane.
Method:
- Sample Preparation: Using a single standard solution for each analyte, pipette different volumes (e.g., 2 mL, 5 mL, 10 mL) into 20 mL headspace vials. This creates phase ratios ((\beta)) of 9, 3, and 1, respectively.
- HS-GC Conditions:
- Use a constant, optimized equilibration temperature (e.g., 70°C).
- Keep all other instrument parameters identical to Protocol 1.
- Analysis: Analyze each sample volume in triplicate. Record and compare peak areas.
Results and Interpretation: The effect of the phase ratio is predictable but highly dependent on the analyte's partition coefficient.
Table 2: Experimental Impact of Phase Ratio (β) on Analyte Response
| Analyte | K Value | Impact of Decreasing β (Increasing Sample Volume) | Theoretical Basis |
|---|---|---|---|
| Ethanol | High (~500) [24] | Negligible increase in headspace concentration [24] | K >> β. The term K dominates the sum (K + β), so changing β has little effect. |
| n-Hexane | Low (~0.01) [24] | Large, proportional increase in headspace concentration [24] | K << β. The phase ratio β dominates. Decreasing β directly decreases the sum (K + β), significantly increasing (C_G). |
For analytes with intermediate K values (~10), the increase in response with sample volume is approximately linear [24]. A standard practice is to use a sample volume that results in a phase ratio of 1 (e.g., 10 mL in a 20 mL vial), which simplifies calculations and often provides a good compromise for analyzing multiple analytes [24].
Static vs. Dynamic Headspace: An Objective Comparison for Solvents Research
While static headspace is robust and widely used, dynamic headspace sampling (DHS) serves as a powerful alternative, especially for challenging applications. The core difference lies in the sampling process, as illustrated below.
The choice between these two techniques depends heavily on analytical goals.
Table 3: Static vs. Dynamic Headspace Comparison for Pharmaceutical Solvents
| Parameter | Static Headspace | Dynamic Headspace (DHS) |
|---|---|---|
| Principle | Equilibrium-based single extraction [21] [27] | Continuous purging and trapping [27] [13] |
| Sensitivity | Good for medium- to high-abundance volatiles [13] | Excellent; superior for trace-level detection due to exhaustive extraction and preconcentration [13] |
| Matrix Effects | Can be significant; complex matrices may retain volatiles, reducing recovery [13] | Can overcome matrix effects via continuous purging; ideal for polar analytes in aqueous matrices and solids [13] |
| Linearity | Good over a limited range | Broader linear dynamic range |
| Throughput & Simplicity | High; simple, easily automated, minimal method development [22] | Lower; requires optimization of purge flow, trap selection, and desorption parameters [13] |
| Ideal Use Case | Routine, high-throughput analysis of residual solvents per USP <467> [23] | Research on trace impurities, profiling complex volatile signatures, and analyzing challenging matrices [3] [13] |
Dynamic headspace is particularly advantageous when static headspace fails to deliver sufficient sensitivity or when dealing with complex matrices that strongly retain volatiles [13]. Advanced DHS variants like the Full Evaporative Technique (FET), where the entire sample is evaporated, can further eliminate matrix effects for certain applications [28] [13].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful headspace method development relies on consistent and high-quality materials.
Table 4: Essential Reagents and Materials for Headspace-GC Research
| Item | Function & Importance | Common Examples / Notes |
|---|---|---|
| Headspace Vials | Container for the sample and headspace gas; must be gas-tight [22] | 10–22 mL vials are common; ensure at least 50% headspace [22] [26] |
| Septa & Caps | Ensures a hermetic seal to prevent loss of volatiles [26] | PTFE/silicone septa with aluminum crimp caps; check temperature limits [26] |
| Diluent | Dissolves or suspends the sample to create a homogeneous matrix [23] | DMA, DMSO, DMF, Water; must be high-purity, low in volatiles, and able to dissolve the sample [23] |
| Salting-Out Agents | Increases ionic strength of aqueous samples, reducing solubility of volatile analytes and driving them into the headspace [24] [26] | Potassium Chloride, Sodium Chloride; most effective for polar analytes [24] |
| GC Inlet Liner | Interface where the headspace sample is vaporized; design affects band broadening and peak shape [26] | Narrow-bore liners (e.g., 1.2 mm ID) recommended for sharper peaks and higher signal [26] |
| Calibration Standards | Critical for accurate quantification; must be matrix-matched to account for K and β [24] [23] | Prepared in the same diluent as samples; internal standard (e.g., n-propanol) improves precision [25] |
For scientists in drug development, mastering the chemical system within a headspace vial is non-negotiable for generating reliable data. The interplay between the partition coefficient (K) and the phase ratio (β) dictates analytical sensitivity. As demonstrated, temperature is a powerful lever for modulating (K), especially for soluble analytes, while the sample volume directly controls (\beta), offering the greatest gains for insoluble volatiles.
The choice between static and dynamic headspace should be guided by the specific analytical challenge. Static headspace remains the workhorse for regulated, high-throughput residual solvents testing due to its robustness and simplicity. However, when methods push into the realm of trace-level analysis, or when confronted with complex, challenging matrices, dynamic headspace sampling provides a potent alternative with superior sensitivity and the ability to overcome significant matrix effects. A deep understanding of these principles empowers researchers to rationally develop, optimize, and validate robust HS-GC methods.
The analysis of volatile organic compounds (VOCs) and residual solvents is critical across pharmaceutical, environmental, and industrial sectors due to toxicity concerns and regulatory requirements. Headspace gas chromatography (HS-GC) has emerged as the premier technique for these analyses, providing a robust means to separate volatile analytes from complex sample matrices without introducing non-volatile interferences. This guide examines key regulatory methods mandating headspace use, specifically the United States Pharmacopeia (USP) <467> protocol for pharmaceutical residual solvents and the Environmental Protection Agency (EPA) methods for environmental contaminants. The analysis is framed within a technical comparison of static (S-HS) versus dynamic headspace (DHS) methodologies, equipping researchers and drug development professionals with the data needed to select appropriate techniques based on sensitivity requirements, regulatory compliance, and analytical efficiency.
Headspace techniques operate by sampling the gaseous phase above a solid or liquid sample in a sealed vial [4]. Static headspace maintains equilibrium conditions where volatiles partition between the sample matrix and the gas phase, while dynamic headspace (also called purge and trap) continuously strips analytes from the sample using an inert gas for preconcentration [3] [4]. The fundamental difference in mechanics leads to significant practical implications for detection capability, sample throughput, and method complexity, making technique selection a critical decision point for regulatory compliance.
Regulatory Framework and Methodologies
USP <467> for Residual Solvents in Pharmaceuticals
USP <467> establishes standardized procedures for determining residual solvents in pharmaceutical drug substances and products, classifying solvents into three categories based on toxicity risk [29]. This method primarily employs static headspace sampling coupled with gas chromatography and flame ionization detection (GC-FID) [30]. The protocol specifies rigorous validation requirements for specificity, sensitivity, precision, and accuracy to ensure patient safety and product quality.
Recent applications demonstrate method development within this framework. A 2025 study developed a validated HS-GC method for determining six residual solvents (methanol, ethyl acetate, isopropyl alcohol, triethylamine, chloroform, and toluene) in losartan potassium raw material [29]. Critical parameters included sample diluent selection (dimethylsulfoxide preferred over water), incubation temperature (100°C), and equilibration time (30 minutes), with chromatographic separation achieved using a DB-624 capillary column [29]. The method demonstrated selectivity, linearity (r ≥ 0.999), and appropriate sensitivity with quantification limits below 10% of ICH specification limits [29].
EPA Methods for Environmental Monitoring
The EPA mandates headspace-based approaches for analyzing volatile organic compounds in various environmental matrices. Key methods include:
- EPA Method 5021A: Provides procedures for equilibrium headspace analysis of VOCs in soils, sediments, and solid wastes in preparation for GC or GC-MS determination [31].
- EPA Method 25E: Determines vapor phase organic concentration in waste samples by analyzing headspace vapor for carbon content using a flame ionization detector [32].
- EPA Method 8260: Allows for VOC analysis in solid waste matrices using either static or dynamic headspace introduction [33].
These methods address environmental monitoring needs with requirements for detection limits, precision, and accuracy in complex sample matrices, with static headspace often employed for prescreening to prevent chromatographic system over-saturation [33].
Static vs. Dynamic Headspace: Technical Comparison
Fundamental Mechanism and Workflow
The core difference between static and dynamic headspace lies in analyte extraction and transfer mechanics. The workflows differ significantly, as illustrated below:
Performance Comparison and Experimental Data
Direct comparative studies provide quantitative performance data between the two techniques. A method comparison study evaluating EPA Method 8260 demonstrated significant differences in detection capability:
Table 1: Static vs. Dynamic Headspace Performance Comparison for EPA Method 8260 VOCs [33]
| Parameter | Static Headspace | Dynamic Headspace |
|---|---|---|
| Detection Limits | 10 ppb | 0.5 ppb |
| Relative Sensitivity | 1x (Baseline) | 20-125x greater |
| Representative Peak Area (10 ppb standard) | 8 kCounts | 200 kCounts |
| Water Management | Moderate water vapor | Efficiently reduced with dry purge |
| Primary Application | Sample prescreening to prevent system saturation | Trace-level analysis and compliance monitoring |
The experimental conditions for this comparison used a Teledyne Tekmar HT3 system with both static and dynamic capabilities, a Varian 431GC 210 MS, and a FactorFour VF-624ms column (20 m × 0.15 mm × 0.84 μm) [33]. The dramatic sensitivity difference, with dynamic headspace providing 20-125 times greater detection depending on the compound, highlights the technique's superior preconcentration capability [33]. For instance, dibromofluoromethane showed 125 times greater peak area with dynamic headspace compared to static at the same concentration [33].
Analytical Characteristics and Application Scope
Beyond sensitivity, the techniques differ significantly in operational parameters and optimal application areas:
Table 2: Operational Characteristics and Application Scope Comparison
| Characteristic | Static Headspace | Dynamic Headspace |
|---|---|---|
| Equilibrium State | Essential (temperature/time controlled) | Not required (continuous extraction) |
| Typical Temperature Range | 45-150°C [3] | Ambient-150°C |
| Analysis Time | Shorter (minutes) | Longer (includes purge/desorb cycles) |
| Automation Potential | High (fully automatic systems available) [34] | Moderate |
| Matrix Effects | Significant (salting out can improve recovery) [3] | Less pronounced due to continuous stripping |
| Preferred Applications | Routine pharmaceutical testing (USP <467>) [30], quality control, high-throughput labs | Environmental trace analysis [33], complex matrices, forensic investigations |
Static headspace excels in pharmaceutical quality control environments where high throughput and simplicity are prioritized over extreme sensitivity. In contrast, dynamic headspace is indispensable for environmental monitoring where regulatory limits often mandate part-per-trillion detection capabilities [33].
Essential Research Reagent Solutions
Successful implementation of headspace methodologies requires specific materials and reagents optimized for volatile compound analysis:
Table 3: Essential Research Reagents and Materials for Headspace Analysis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Dimethylsulfoxide (DMSO) | Sample diluent | High boiling point (189°C) minimizes interference; preferred over water for certain pharmaceutical applications [29] |
| DB-624/VR-1/VR-5 GC Columns | Chromatographic separation | Standard stationary phases for volatile compounds; used in both USP and EPA methods [29] [30] |
| Polydimethylsiloxane (PDMS) Fiber | SPME extraction | Non-polar coating for SPME-based static headspace; excellent for hydrocarbons [3] |
| Divinylbenzene/PDMS (DVB/PDMS) Fiber | SPME extraction | Bipolar coating expands analyte range; common for flavor and fragrance compounds [3] |
| Helium Carrier Gas | Chromatographic mobile phase | Provides optimal separation efficiency; used in most regulatory methods [29] |
| Certified Reference Standards | Quantification and calibration | Essential for method validation; concentrations should trace to primary standards |
The choice between static and dynamic headspace methodologies depends primarily on sensitivity requirements, sample throughput needs, and matrix complexity. Static headspace provides a robust, easily automated solution for pharmaceutical quality control (USP <467>) and other applications where target compounds are present at concentrations ≥10 ppb. Its simplicity, speed, and compatibility with fully automated systems make it ideal for high-volume testing environments. Conversely, dynamic headspace (purge and trap) offers superior sensitivity for trace-level analysis (sub-ppb detection) required in environmental monitoring (EPA methods) and specialized applications, albeit with increased method complexity and analysis time.
The expanding regulatory landscape continues to drive headspace technology innovation, with current trends focusing on enhanced automation, improved sensitivity, and method harmonization across industries [34]. Understanding the technical capabilities and limitations of each approach enables researchers to select the optimal technique for specific regulatory requirements and analytical challenges.
Selecting and Applying Headspace Methods: From USP Compliance to Trace Analysis
In the analytical scientist's toolkit, static headspace gas chromatography (HS-GC) stands as a technique of choice for volatile compound analysis across pharmaceutical, food, and environmental sectors. As a standardized approach for routine quality control, static headspace provides a robust, efficient, and matrix-sparing solution for analyzing volatile organic compounds without introducing complex sample matrices into the chromatographic system. This technique operates on the principle of equilibrium partitioning, where volatile compounds distribute between the sample matrix and the gas phase (headspace) in a sealed vial under controlled temperature conditions. A portion of this headspace is then injected into the GC system for analysis [12] [35].
For researchers and drug development professionals navigating the choice between static and dynamic headspace techniques, understanding the specific applications where static headspace excels is crucial for method development and laboratory efficiency. While dynamic headspace (purge and trap) offers superior sensitivity for trace-level analysis through continuous purging and trapping of volatiles [12] [36], static headspace provides distinct advantages in simplicity, reproducibility, and cost-effectiveness for numerous routine applications. This guide examines the technical foundations, applications, and experimental protocols that establish static headspace as an indispensable tool for specific analytical scenarios.
Technical Comparison: Static vs. Dynamic Headspace
Fundamental Operational Differences
The core distinction between static and dynamic headspace lies in their sampling mechanisms and concentration capabilities, which directly influence their appropriate application domains.
Table 1: Core Technical Differences Between Static and Dynamic Headspace
| Feature | Static Headspace GC | Dynamic Headspace GC |
|---|---|---|
| Principle | Equilibrium-based sampling | Continuous purging with inert gas |
| Sensitivity | Good for many volatiles (typically ppm-ppb) | Higher sensitivity for trace-level analysis (ppb-ppt) |
| Analysis Time | Longer equilibration time required | Generally faster analysis |
| Complexity | Simpler setup and operation | More complex setup with trapping |
| Sample Preparation | Minimal preparation required | Requires setup for gas flow and trapping |
| Risk of Contamination | Lower risk due to closed system | Potential for loss of volatiles or system contamination |
Quantitative Performance Metrics
A systematic comparison of headspace techniques reveals distinct performance characteristics that inform method selection. Research demonstrates that while static sampling techniques exhibit sufficient extraction yields (approximately 10-20%) to be reliably used down to approximately 100 ng/L, enrichment techniques (including dynamic headspace) display extraction yields of up to 80%, resulting in method detection limits (MDLs) down to the picogram per liter range [16]. The relative standard deviations (RSDs) for static headspace are typically below 10%, and frequently at or below 2.0% with modern automated systems, comparable to precision found with liquid-chromatography injection [35].
Table 2: Quantitative Performance Comparison of Headspace Techniques
| Technique Class | Extraction Yield | Method Detection Limits | Precision (RSD) |
|---|---|---|---|
| Static Sampling | 10-20% | ~100 ng/L | <10%, frequently ≤2.0% |
| Static Enrichment | Up to 80% | Picogram per liter range | Below 27% |
| Dynamic Enrichment | Up to 80% | Picogram per liter range | Below 27% |
Key Application Domains for Static Headspace
Residual Solvents in Pharmaceuticals
Static headspace sampling has become the standard technique for determining residual solvents in pharmaceutical drug substances according to pharmacopoeia requirements (USP <467>, European Pharmacopoeia) [37] [35]. The technique's ability to efficiently detect and quantify Class 1, 2, and 3 solvents in active pharmaceutical ingredients (APIs) without matrix interference makes it indispensable for quality control laboratories. A validated static headspace method for ethanol, tetrahydrofuran, and toluene determination using water-dimethylformamide mixture as sample solvent demonstrates excellent sensitivity, precision, and recovery, meeting ICH validation guidelines [37].
The pharmaceutical industry's stringent regulatory requirements for drug safety and efficacy have established static headspace as a cornerstone technique for quality control. The accuracy and efficiency offered by static headspace in detecting volatile impurities in drug formulations make it an essential tool in pharmaceutical manufacturing and quality assurance processes [38].
Flavor and Fragrance Analysis
In food and beverage quality control, static headspace provides an effective solution for detecting volatile compounds that affect flavor, aroma, and shelf-life [12] [38]. The technique is particularly valuable for routine analysis of flavor compounds in products where the volatile profile represents key quality attributes. While dynamic headspace may offer greater sensitivity for trace aroma compounds, static headspace delivers sufficient sensitivity for many quality control applications with significantly simpler operation and faster turnaround times.
The technique's minimal sample preparation and clean injections make it ideal for complex food matrices, protecting chromatographic columns from non-volatile components while providing representative volatile profiles. This capability is crucial for maintaining product standards and compliance with food safety regulations as consumer awareness regarding food quality continues to increase [38].
Routine Quality Control Applications
The operational simplicity and reproducibility of static headspace make it particularly suited for high-throughput quality control laboratories where robustness and ease of use are paramount. Key characteristics that favor static headspace in routine QC include:
- Minimal analyst intervention with automated systems
- Reduced contamination risk from non-volatile matrix components
- Excellent precision with RSD consistently below 10%
- Lower operational complexity compared to dynamic systems
- Cost-effectiveness for routine testing environments
These advantages explain why static headspace currently holds a significant portion of the headspace samplers market, though dynamic headspace is anticipated to grow at a faster rate as detection requirements become more stringent [38].
Experimental Protocols and Method Optimization
Standard Static Headspace Methodology
A typical static headspace analysis follows a standardized workflow that ensures reproducible results across various applications. The process begins with sample preparation, where the sample is weighed or measured into a headspace vial, often with an appropriate diluent. The vial is immediately sealed with a septum cap to prevent volatile loss. The sealed vial is then transferred to the headspace sampler oven, where it undergoes controlled heating at a specified temperature for a predetermined equilibration time. During this phase, volatile compounds partition between the sample matrix and the headspace. Once equilibrium is established, the instrument pressurizes the vial with carrier gas, and a portion of the headspace vapor is automatically transferred to the GC inlet for chromatographic separation and detection [35].
Critical Method Parameters and Optimization
Several experimental parameters significantly impact static headspace performance and require systematic optimization during method development:
Sample Volume Optimization The relationship between sample volume and analytical response in static headspace is non-linear due to the fixed vial volume. Research demonstrates that increasing sample volume beyond an optimal point (typically 1-2 mL in a 20-mL vial) does not yield expected signal increases and may actually decrease response for some analytes [35]. This counterintuitive behavior stems from the reduced headspace volume available for vapor partitioning, highlighting the need for empirical optimization rather than theoretical assumptions.
Equilibration Time Equilibration time represents a critical parameter balancing analysis speed and sensitivity. Studies comparing incubation times of 5, 10, 15, 30, and 60 minutes demonstrate that times longer than 5-10 minutes often do not yield significant signal increases for many volatile compounds [35]. This finding contrasts with some pharmacopeial recommendations of 60-minute equilibration, suggesting potential for significant time savings in routine analysis without compromising data quality.
Diluent Composition The choice of diluent significantly influences volatile partitioning into the headspace. For aqueous samples, the addition of salts can enhance sensitivity through salting-out effects, while for challenging matrices, high-boiling solvents like dimethyl sulfoxide (DMSO) or dimethylformamide (DMF) may improve recovery [37] [35]. Systematic evaluation of diluent composition represents a powerful approach for optimizing method sensitivity, particularly for problematic analytes.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Static Headspace Analysis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Water-DMF Mixture | Sample solvent for residual solvents | Provides good sensitivity and recovery for pharmaceutical compounds [37] |
| DMSO | High-boiling diluent | Enhances volatility of analytes for improved headspace partitioning [35] |
| Salts (e.g., NaCl) | Salting-out agent | Increases volatile compound transfer to headspace in aqueous matrices [35] |
| Internal Standards | Quantitation reference | Corrects for vial-to-vial variation; deuterated analogs preferred for MS detection |
| Headspace Vials | Sample containment | Certified vials with PTFE-faced silicone septa prevent volatile loss and contamination |
Decision Framework: When to Select Static Headspace
Ideal Use Cases for Static Headspace
Based on comparative performance data and application requirements, static headspace represents the optimal choice when:
- Analyzing relatively concentrated volatile compounds (typically ppm level) where extreme sensitivity is not required [12] [16]
- Operating in regulatory environments with established static headspace methods (e.g., USP <467>) [37] [35]
- Prioritizing method robustness and simplicity in high-throughput quality control laboratories [12] [38]
- Working with complex matrices where minimal sample preparation is desirable to protect instrumentation [12] [35]
- Managing resource constraints where operational simplicity and cost-effectiveness are paramount [38]
When to Consider Dynamic Headspace Alternatives
Dynamic headspace techniques (including purge and trap and ITEX systems) become preferable when analytical requirements include:
- Ultra-trace analysis demanding detection limits in the parts-per-trillion range [12] [16]
- Complex volatile profiles requiring exhaustive extraction for comprehensive characterization [36] [4]
- Sample types with strong matrix binding of volatiles that benefit from continuous purging [12] [36]
- Research applications where maximum sensitivity justifies increased method complexity and cost [38]
Static headspace gas chromatography remains an indispensable analytical technique with particular strength in pharmaceutical residual solvents testing, flavor and fragrance analysis, and routine quality control applications. Its equilibrium-based approach delivers sufficient sensitivity for many regulatory requirements while offering operational simplicity, excellent precision, and minimal sample preparation. While dynamic headspace methods provide superior sensitivity for trace-level analysis, the practical advantages of static headspace in robustness, cost-effectiveness, and regulatory acceptance secure its position as a fundamental tool in analytical laboratories. By understanding the technical principles, optimization strategies, and application boundaries outlined in this guide, researchers and quality control professionals can make informed decisions about when static headspace provides the optimal solution for their volatile compound analysis needs.
For researchers in drug development and analytical science, the analysis of volatile impurities is a critical component of product safety and quality control. Within this field, headspace gas chromatography (HS-GC) has emerged as a powerful technique for analyzing volatile compounds in complex matrices. However, a fundamental choice confronts the scientist: whether to employ static or dynamic headspace sampling. While static headspace is widely valued for its simplicity and robustness, dynamic headspace provides a specialized approach for challenging analytical scenarios where superior sensitivity and trace-level detection are paramount [12] [4].
Static headspace operates on an equilibrium-based principle, where a sample is heated in a sealed vial until the volatile compounds partition between the sample matrix and the gas phase above it. A portion of this headspace gas is then injected into the GC system [12] [35]. In contrast, dynamic headspace (also referred to as purge and trap) is a continuous extraction technique. It involves purging the sample with an inert gas, which sweeps the volatile compounds from the sample matrix and onto a focusing trap. The trapped analytes are then thermally desorbed and transferred to the GC column for analysis [12] [4] [39]. This fundamental difference in methodology underlies their distinct performance characteristics and application suitability, which this guide will explore in detail.
Principles and Performance: A Technical Comparison
The operational differences between static and dynamic headspace sampling lead directly to their contrasting performance profiles. The following table summarizes the key technical characteristics of each technique.
Table 1: Fundamental Characteristics of Static and Dynamic Headspace
| Feature | Static Headspace | Dynamic Headspace |
|---|---|---|
| Core Principle | Equilibrium-based sampling [12] | Continuous purging with inert gas [12] |
| Sensitivity | Good for many volatiles [12] | Higher sensitivity for trace-level analysis [12] [38] |
| Analyte Transfer | Single aliquot injection [12] | Continuous stripping and trapping of analytes [12] [39] |
| Typical Applications | Residual solvents, flavors, VOCs in simple matrices [12] [37] [23] | Trace volatiles in water, air, solids; complex matrices [12] [4] |
| Complexity & Cost | Simpler setup, more cost-effective [12] [38] | More complex setup, requires trapping equipment [12] [2] |
The defining advantage of dynamic headspace is its superior pre-concentration capability. While static headspace injects only a single aliquot of the equilibrium headspace, dynamic headspace continuously strips volatiles over time, focusing them onto a trap. This process allows for the injection of a much larger fraction of the total analytes present in the sample, resulting in significantly lower detection limits [12] [39]. This makes it indispensable for detecting impurities at parts-per-billion (ppb) or even sub-ppb levels, such as nitrosamines in pharmaceuticals [40].
However, this enhanced sensitivity comes with trade-offs in operational complexity. Dynamic headspace requires additional equipment, including a gas flow control system for purging and a specialized trap for analyte focusing [12] [2]. The method development process can also be more involved, requiring optimization of purge time, trap selection, and desorption parameters. Consequently, static headspace remains the preferred choice for routine analyses where its simplicity, robustness, and lower cost offer significant practical advantages, especially in regulated environments like pharmaceutical quality control labs performing compendial methods [23].
Decision Framework: When Dynamic Headspace is the Optimal Choice
The choice between static and dynamic headspace is not a matter of one technique being universally superior, but rather of selecting the right tool for the specific analytical challenge. The following workflow diagram provides a logical pathway for making this critical methodological choice.
Diagram 1: A logical workflow for selecting between static and dynamic headspace techniques.
Based on this decision framework, dynamic headspace becomes the unequivocal choice in several key scenarios:
Analysis of Trace-Level Impurities: When method requirements call for detection limits at the ppb or sub-ppb level, dynamic headspace is often the only viable option. Its pre-concentration capability is crucial for detecting potent carcinogens like N-nitrosodimethylamine (NDMA) in pharmaceuticals, where acceptable intake levels can be in the nanogram per day range [39] [40]. One study on nitrosamines in metformin achieved limits of detection of 0.05-0.51 ng/mL using a headspace GC-IMS method, demonstrating the sensitivity required for such applications [40].
Complex or Solid Matrices: For solid samples such as polymers, gels, or powdered tablets, it can be difficult or impossible to prepare matrix-matched calibration standards for static headspace. Dynamic headspace's continuous purging action is more effective at extracting volatiles from these challenging matrices [39]. This is particularly valuable in pharmaceutical packaging studies, where quantifying volatile migrants from polymeric materials is essential [39].
Analysis of Semi-Volatile Compounds: While static headspace is excellent for highly volatile compounds, its sensitivity drops significantly for semi-volatiles with higher boiling points. The continuous extraction in dynamic headspace and the use of heated transfer lines (up to 300°C) make it suitable for a wider range of volatile and semi-volatile compounds [2].
Experimental Protocols and Data Output
To illustrate the practical implementation and output of dynamic headspace, we can examine its application in the analysis of nitrosamines in pharmaceutical products and volatile organic compounds (VOCs) in environmental samples.
Table 2: Exemplary Experimental Protocol for Nitrosamine Analysis via Dynamic Headspace
| Parameter | Specification | Rationale & Impact |
|---|---|---|
| Sample Preparation | Powdered tablets directly weighed into headspace vial [40]. | Minimizes sample preparation, avoids complex extraction. |
| Incubation Temperature | 60-80°C [40]. | Optimizes release of nitrosamines from matrix without degradation. |
| Incubation Time | 10-15 minutes [40]. | Balances efficient extraction with reasonable cycle time. |
| Purge Gas & Flow | Inert gas (N₂ or He), typically 10-60 mL/min [12] [4]. | Strips volatiles from sample; flow rate affects recovery. |
| Trap Type | Multi-bed sorbent (e.g., Tenax, charcoal, carbon molecular sieves) [2]. | Ensures efficient trapping of a wide volatility range. |
| Desorption Temperature | 180-300°C [2]. | Must be sufficient to rapidly transfer analytes to GC column. |
The quantitative performance of a well-optimized dynamic headspace method is demonstrated in a study for seven nitrosamine impurities, yielding the following results [40]:
Table 3: Quantitative Performance Data for Nitrosamine Analysis via HS-GC-IMS
| Analyte | Linear Range (ng/mL) | R² | LOD (ng/mL) | LOQ (ng/mL) | Precision (Intra-day RSD) |
|---|---|---|---|---|---|
| NDMA | 0.5 - 100 | >0.99 | 0.05 | 0.16 | < 5% |
| NDEA | 0.5 - 100 | >0.99 | 0.11 | 0.36 | < 5% |
| NPYR | 0.5 - 100 | >0.99 | 0.51 | 1.70 | < 5% |
| Other Nitrosamines | 0.5 - 100 | >0.99 | 0.06 - 0.51 | 0.19 - 1.70 | < 5% |
The data in Table 3 confirms that dynamic headspace-based methods can achieve the necessary sensitivity, wide linear dynamic range, and high precision required for modern pharmaceutical impurity analysis. The technique's effectiveness is further highlighted in applications involving complex matrices, such as the use of Multiple Headspace Extraction (MHE) for quantifying formaldehyde in a Gelucire excipient or styrene in polystyrene polymers, where matrix-matched calibration is problematic [39].
The Scientist's Toolkit: Essential Reagents and Materials
Implementing a robust dynamic headspace method requires careful selection of consumables and reagents. The following table details the essential components of a dynamic headspace workflow.
Table 4: Essential Research Reagent Solutions for Dynamic Headspace Analysis
| Item | Function & Importance | Technical Specifications & Selection Criteria |
|---|---|---|
| Sorbent Trap | Focuses analytes stripped during purging; heart of the system [2]. | Multi-bed configurations (e.g., Tenax, charcoal, molecular sieve) provide a broad analyte range. Must be inert and thermally stable. |
| High-Purity Inert Gas | Purge gas for transferring volatiles from sample to trap [12]. | Helium or Nitrogen (99.999% purity). Must be oxygen-free to prevent analyte degradation. |
| Headspace Vials | Contain the sample in a sealed, inert environment [12]. | 10-20 mL volume, clear glass. Must be compatible with autosampler. |
| Septa & Caps | Maintain a leak-proof seal during heating and pressurization [41]. | PTFE-faced silicone septa, aluminum crimp caps. High-temperature stability is critical. |
| High-Boiling Solvent | Sample diluent for improving analyte release [23] [35]. | DMF, DMSO, DMA. Must dissolve sample, have low vapor pressure, and not interfere. |
Dynamic headspace analysis stands as a powerful, specialized technique within the analytical chemist's arsenal, uniquely suited to overcoming the challenges of trace-level volatile impurity analysis in complex solid matrices. While static headspace remains the workhorse for routine analysis due to its operational simplicity, dynamic headspace is the unequivocal choice when method requirements push the boundaries of sensitivity, particularly for potent impurities like nitrosamines in pharmaceuticals or VOCs in environmental samples [12] [40].
The future of dynamic headspace is closely tied to technological advancements aimed at increasing throughput and simplifying operation. The integration of automated, high-speed sampling systems and advanced detection techniques like Selected Ion Flow Tube Mass Spectrometry (SIFT-MS) is already transforming MHE into a more cost-effective, routine approach [39]. Furthermore, the ongoing development of novel miniaturized devices for dynamic headspace sampling promises to enhance sensitivity while reducing sample and solvent consumption, aligning with the broader goals of green analytical chemistry [4]. For researchers and drug development professionals, mastering the application of dynamic headspace is not merely an academic exercise, but a critical competency for ensuring product safety and regulatory compliance in an increasingly demanding analytical landscape.
Headspace sampling is a premier technique in gas chromatography (GC) for analyzing volatile organic compounds in complex sample matrices. By analyzing the gas phase above a sample, it effectively minimizes the introduction of non-volatile contaminants into the GC system, thereby enhancing analytical robustness and instrument uptime [12] [42]. This guide provides a detailed comparison of the two principal headspace methodologies—static and dynamic headspace—focusing on their application across pharmaceutical, food, and environmental matrices. The selection between these techniques is often dictated by the nature of the analyte, the complexity of the sample matrix, and required sensitivity levels, with regulatory requirements frequently providing definitive guidance [21].
Static headspace gas chromatography (S-HS-GC) is an equilibrium-based technique where the sample is placed in a sealed vial and heated to allow volatile compounds to partition between the sample matrix and the gas phase above it [12] [21]. After equilibration, a portion of this headspace gas is injected into the GC system. In contrast, dynamic headspace gas chromatography (D-HS-GC), often referred to as purge-and-trap, involves continuously purging the sample with an inert gas, which sweeps volatile compounds from the sample onto a concentrating trap. The trapped analytes are subsequently thermally desorbed into the GC for analysis [12] [4]. This fundamental operational difference—equilibrium versus exhaustive extraction—underpins their distinct performance characteristics and suitability for different analytical challenges.
Fundamental Principles and Comparative Performance
Theoretical Foundations
The theoretical foundation of static headspace is described by the equilibrium relationship between the sample and its gas phase. The detector response (A) is proportional to the gas phase concentration (CG), which is determined by the original sample concentration (C0), the partition coefficient (K), and the phase ratio (β) as defined by the equation: A ∝ CG = C0 / (K + β) [42] [21].
The partition coefficient (K = CS/CG) represents the ratio of an analyte's concentration in the sample phase (CS) to its concentration in the gas phase (CG) at equilibrium and is strongly influenced by temperature and sample composition [21]. The phase ratio (β = VG/VS) is the ratio of the gas phase volume (VG) to the sample phase volume (VS) within the sealed vial [42] [21]. To maximize detector response, the sum of K and β must be minimized, which is typically achieved through optimization of temperature, sample volume, and vial size [42]. For compounds with high solubility in the sample matrix (K >> β), even minor temperature fluctuations can significantly impact results, whereas for low-solubility compounds (K << β), the impact of temperature variation is less pronounced [21].
Dynamic headspace operates on a different principle, as it does not rely on establishing a static equilibrium. Instead, it employs continuous purging to exhaustively transfer volatiles from the sample to the trapping medium [12] [4]. This continuous displacement of the headspace creates a concentration gradient that continually drives the release of analytes from the sample matrix, effectively overcoming the equilibrium limitations of static headspace and enabling complete extraction of trace-level volatiles [43].
Comparative Performance Data
The following table summarizes the key operational and performance characteristics of static versus dynamic headspace techniques, highlighting their distinct advantages and limitations for different analytical scenarios.
Table 1: Performance Comparison of Static vs. Dynamic Headspace GC
| Feature | Static Headspace GC | Dynamic Headspace GC |
|---|---|---|
| Fundamental Principle | Equilibrium-based sampling [12] | Continuous purging with inert gas [12] |
| Typical Sensitivity | Good for many volatiles [12] | Higher sensitivity, suitable for trace-level analysis [12] [43] |
| Analysis Time | Longer due to required equilibration time [12] | Generally faster analysis [12] |
| Sample Preparation | Minimal preparation required [12] | Requires setup for gas flow and trapping [12] |
| System Complexity | Simpler setup [12] | More complex, requires traps and gas flow systems [12] |
| Risk of Contamination | Lower risk due to closed system [12] | Potential for loss of volatiles if not controlled [12] |
| Optimal Use Cases | Relatively simple matrices; volatile compounds [12] [44] | Trace analysis in complex matrices; low-volatility compounds [12] [45] |
Experimental Protocols and Methodologies
Standard Static Headspace Protocol for Residual Solvents
The United States Pharmacopeia (USP) method <467> for residual solvents in pharmaceuticals represents a well-established application of static headspace GC [42] [23]. A generic methodology can be applied for the determination of common Class 2 and Class 3 solvents [23].
Sample Preparation: Accurately weigh approximately 100 mg of the active pharmaceutical ingredient (API) into a 20-mL headspace vial. For solid dosage forms, crush tablets to a fine powder or carefully open capsules to obtain a representative sample. Add 1 mL of a suitable high-boiling-point diluent such as dimethylacetamide (DMA) or dimethyl sulfoxide (DMSO) to the vial. Seal the vial immediately with an aluminum crimp cap equipped with a polytetrafluoroethylene (PTFE)-lined septum. Vortex the vial gently to ensure complete dissolution or homogeneous suspension of the sample [23].
Instrumental Conditions:
- GC Column: 30 m × 0.32 mm ID, 1.8 µm df mid-polarity column (e.g., 6% cyanopropylphenyl/94% dimethylpolysiloxane equivalent to USP G43).
- Carrier Gas: Helium or Nitrogen, constant flow of 1.5 mL/min.
- Oven Program: Initial temperature 40°C (hold 5 min), ramp to 240°C at 15-20°C/min.
- Headspace Sampler: Equilibration temperature: 80-120°C; Equilibration time: 20-60 min; Loop temperature: 110-130°C; Transfer line temperature: 120-140°C [23].
Quantitation: Prepare a multi-component stock standard solution in the same diluent used for samples. Use external standardization, comparing peak areas from sample vials to those from standard solutions, corrected for sample weight [23].
Dynamic Headspace (Purge & Trap) Protocol for Water Analysis
U.S. Environmental Protection Agency (USEPA) Method 524.2 for the analysis of volatile organic compounds (VOCs) in drinking water is a canonical application of dynamic headspace GC-MS [21].
Sample Collection and Preservation: Collect water samples in 40-mL glass vials with zero headspace, sealed with TFE-faced silicone septa. Preserve with hydrochloric acid to pH <2 and store at 4°C until analysis [21].
Purge-and-Trap Conditions:
- Purge Gas: High-purity helium at a flow rate of 20-40 mL/min.
- Purge Time: 10-15 minutes at ambient temperature.
- Purge Device: A fritted glass purger is commonly used to create fine gas bubbles for efficient stripping of VOCs.
- Trap: A multi-bed adsorbent trap, typically containing Tenax, silica gel, and activated charcoal.
- Dry Purge: 1-2 minutes with helium to remove residual water vapor from the trap.
- Desorption: Thermal desorption at 180-250°C for 2-4 minutes into the GC inlet. Backflush the trap during desorption to prevent high-boiling compounds from entering the GC system.
GC-MS Conditions:
- Column: 60 m × 0.25 mm ID, 1.4 µm film thickness DB-624 or equivalent.
- Oven Program: 35°C (hold 5 min) to 220°C at 6-8°C/min.
- MS Detection: Full scan mode (e.g., m/z 35-300) with electron ionization (EI).
- Calibration: Internal standard method using deuterated or fluorinated analogs of target analytes (e.g., toluene-d8, 4-bromofluorobenzene) [21].
Workflow Diagrams
Static Headspace Workflow
Dynamic Headspace Workflow
Matrix-Specific Considerations and Applications
Pharmaceutical Analysis
Pharmaceutical analysis demands robust, reproducible methods for quality control, with residual solvent testing being a primary application. Static headspace is the mandated technique for USP <467> due to its simplicity and reliability for quality control environments [42] [23]. The selection of an appropriate diluent is critical; water, DMSO, DMA, and DMF are commonly used, with the choice dependent on the drug substance's solubility and stability [44] [23]. For instance, DMSO is often preferred over DMF due to its lower toxicity and higher boiling point [44]. Method development must account for the partition coefficient (K), which can be optimized by adjusting temperature, sample volume, and vial size to ensure adequate sensitivity for all target solvents [21] [23].
For challenging pharmaceutical applications, such as analyzing very low volatility compounds or those with high affinity for the matrix, dynamic headspace or specialized techniques like the Full Evaporative Technique (FET) may be necessary. The FET uses a very small sample volume (e.g., <100 µL) in a large vial (10-20 mL) and employs elevated temperatures to completely vaporize the sample, effectively eliminating the matrix effect and improving the release of less volatile analytes [43].
Food and Beverage Analysis
Food matrices present unique challenges due to their diversity, complexity, and non-homogeneous nature [45]. Headspace techniques are extensively used for analyzing flavors, aromas, and contaminants like furan or chloropropanols [45].
Static headspace is ideal for quality control of highly volatile compounds in foods, such as monitoring ethanol content or residual packaging solvents [44]. For more complex aroma profiles or trace-level contaminants, dynamic headspace or sorptive extraction techniques like Solid-Phase Microextraction (SPME) offer superior performance [45] [4]. Dynamic headspace provides a more comprehensive extraction of the volatile profile from solid foods, as demonstrated in the analysis of dry tea, where it detected a wider range of compounds with greater sensitivity compared to static headspace [43].
The phase ratio (β) is a critical parameter in food analysis. Using a larger vial (20 mL vs. 10 mL) with the same sample volume decreases β, which can significantly increase the concentration of analytes in the headspace and improve detection limits [42]. Temperature control is equally vital, as increasing the equilibration temperature decreases the partition coefficient (K) for many analytes, driving more compound into the headspace and enhancing sensitivity, though care must be taken to avoid analyte degradation or generating process artifacts [42].
Environmental Analysis
Environmental monitoring of water, soil, and air requires methods with high sensitivity and specificity, often mandated by regulatory agencies. Dynamic headspace (purge-and-trap) is the benchmark technique for the analysis of VOCs in water, as specified in USEPA Method 524.2, due to its exceptional sensitivity at parts-per-trillion levels [21]. The continuous purging action efficiently extracts volatile compounds from the aqueous matrix, while the trapping and thermal desorption steps provide significant preconcentration, enabling the detection of ultratrace contaminants [21] [4].
Static headspace finds application in environmental analysis for simpler screening purposes or for samples with high fouling potential where dynamic systems might be compromised [46]. It is also suitable for the analysis of VOCs in soil samples, though careful attention must be paid to achieving equilibrium from solid matrices, which may require longer equilibration times or the addition of a modifier like water or solvent to facilitate the release of analytes [21]. For complex environmental matrices, Multiple Headspace Extraction (MHE) can be employed to achieve quantitative extraction by performing successive static extractions from the same vial until the analyte is exhausted, which is particularly useful for soils with strong adsorption characteristics [46].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful headspace analysis requires careful selection of consumables and reagents to ensure method accuracy, precision, and sensitivity.
Table 2: Essential Materials for Headspace Analysis
| Item | Function & Importance | Selection Considerations |
|---|---|---|
| Headspace Vials | Contain sample in a sealed, inert environment for heating and pressurization [42]. | Common sizes are 10 mL and 20 mL; choice affects phase ratio (β) [42]. |
| Septa & Caps | Provide a gas-tight seal to prevent loss of volatile analytes during incubation [42]. | Must have PTFE/silicone facing for high-temperature stability and chemical inertness [23]. |
| Diluent | Dissolves or suspends the sample to facilitate release of volatiles [44] [23]. | High boiling point (e.g., DMSO, DMA, DMF); must not interfere with analysis [23]. |
| Internal Standards | Correct for variability in sample preparation and injection [45]. | Deuterated or structurally similar analogs not present in the native sample [45]. |
| Adsorbent Tubes/Traps | (D-HS) Trap and concentrate volatiles purged from the sample [12] [4]. | Packing (e.g., Tenax, Carbopack) is selected based on the volatility range of target analytes [4]. |
| Gas Tight Syringe | (Manual S-HS) Transfer the headspace aliquot from vial to GC injector [44]. | Heated to prevent condensation of analytes during transfer [44]. |
The choice between static and dynamic headspace sampling is fundamentally a balance between analytical requirements and practical constraints. Static headspace GC offers a straightforward, robust, and easily automated solution for the analysis of volatile compounds in relatively simple matrices, making it the workhorse for pharmaceutical quality control labs operating under USP <467> [12] [23]. Its simplicity, minimal sample preparation, and lower risk of contamination are significant advantages for routine analysis.
Dynamic headspace GC, with its superior sensitivity and exhaustive extraction capabilities, is indispensable for trace-level analysis in complex matrices such as environmental waters and for comprehensive profiling of food aromas and contaminants [12] [45] [4]. While it demands a more complex instrumental setup and method optimization, its ability to detect compounds at ultratrace levels often makes it the only viable option for stringent regulatory monitoring and advanced research applications. Ultimately, the selection should be guided by the specific analytical problem, considering the nature of the analyte, the complexity of the matrix, required detection limits, and any governing regulatory methods.
In pharmaceutical research and drug development, the analysis of residual solvents is a critical quality control step, governed by strict pharmacopeial standards such as the United States Pharmacopeia (USP) chapter <467> [47]. Gas Chromatography (GC) is the cornerstone technique for this analysis, and headspace sampling is the preferred method for introducing volatile analytes into the GC system while preventing non-volatile matrix components from contaminating the inlet and column [48]. The two principal headspace techniques are static headspace and dynamic headspace (often referred to as Purge and Trap). The choice between them significantly impacts method sensitivity, throughput, complexity, and its fitness for a given application. This guide provides an objective comparison of these techniques, focusing on their instrumentation, setup, and experimental performance data to inform scientists configuring their GC systems for solvent research.
Core Principles and Instrumentation
Static Headspace Gas Chromatography
In static headspace GC, the sample is placed in a sealed vial and heated to a controlled temperature, allowing volatile compounds to partition between the sample matrix and the gas phase (headspace) until equilibrium is reached [12] [48]. Once equilibrium is achieved, a portion of the headspace vapor is automatically transferred to the GC inlet for analysis.
Key Instrumental Components:
- Sealed Vial: A gas-tight vial (e.g., standard 22-mL headspace vial) that contains the sample.
- Temperature-Controlled Agitator: An incubator that heats the vial to a precise temperature to promote the release of volatiles.
- Gas-Tight Syringe: A heated syringe that extracts a defined volume of the headspace vapor and injects it into the GC inlet.
The fundamental relationship governing the concentration of an analyte in the headspace ((CG)) is derived from its original concentration in the sample ((C0)), the partition coefficient ((K)), and the phase ratio ((\beta)), where (K) is the ratio of the analyte's concentration in the liquid phase to that in the gas phase ((K = CS / CG)), and (\beta) is the ratio of the gas volume to the sample volume ((VG / VS)) in the vial [48]. The equation is expressed as: ( CG = C0 / (K + \beta) ) This relationship shows that for soluble analytes (large (K)), the gas-phase concentration is highly dependent on temperature, while for insoluble volatiles (small (K)), the phase ratio is more influential [48].
Dynamic Headspace Gas Chromatography
Dynamic headspace GC involves continuously purging the sample with an inert gas (e.g., helium or nitrogen). This gas stream sweeps the volatile compounds from the sample and carries them to a trap, where the analytes are adsorbed and concentrated. After the purging cycle, the trap is rapidly heated to desorb the trapped volatiles, which are then transferred to the GC column for analysis [12].
Key Instrumental Components:
- Purge Gas System: A controlled source of inert gas that flows through or over the sample.
- Adsorptive Trap: A tube packed with one or more adsorbent materials (e.g., Tenax, charcoal, silica) to capture and concentrate the volatiles.
- Thermal Desorption Unit: A system that rapidly heats the trap to release the concentrated analytes into the GC inlet.
The following workflow diagram illustrates the core steps and instrumental setup for both static and dynamic headspace techniques.
Comparative Performance and Experimental Data
Direct Comparison of Static Headspace Techniques
A head-to-head study compared traditional GC-FID with a direct-injection mass spectrometry technique (SIFT-MS) for analyzing residual solvents in pharmaceutical products, using static headspace sampling for both [47]. The following table summarizes key performance metrics from this study.
Table 1: Performance Comparison of GC-FID and SIFT-MS for Residual Solvent Analysis [47]
| Performance Metric | GC-FID (USP <467>) | SIFT-MS (Alternative) |
|---|---|---|
| Linearity (R²) | > 0.94 | > 0.97 |
| Repeatability (%RSD) | < 17% | < 10% |
| Analysis Time | ~60 minutes | ~5.4 minutes (11x faster) |
| Accuracy/Recovery | Passed for most compounds (some failures) | Passed for most compounds (fewer failures) |
| Daily Sample Throughput | Lower | 11-fold higher |
This data demonstrates that while both static headspace methods can meet validation criteria, the SIFT-MS technique, which forgoes chromatographic separation, offered significant improvements in analysis speed and throughput without compromising data quality [47].
General Comparison of Static vs. Dynamic Headspace
The fundamental differences in the principles of static and dynamic headspace lead to distinct performance characteristics, making each suitable for different analytical scenarios.
Table 2: General Characteristics of Static vs. Dynamic Headspace GC [12]
| Feature | Static Headspace GC | Dynamic Headspace GC |
|---|---|---|
| Principle | Equilibrium-based sampling | Continuous purging with inert gas |
| Sensitivity | Good for many volatiles | Higher sensitivity, suitable for trace-level analysis |
| Sample Preparation | Minimal preparation required | Requires setup for gas flow and trapping |
| Analysis Time | Longer due to equilibration time | Generally faster analysis |
| Applications | Residual solvents, flavors, VOCs in pharmaceuticals and food [47] [49] | Trace volatiles in water, air, and solid samples [12] |
| System Complexity | Simpler setup | More complex setup (traps, gas flow) |
Efficiency of Different Extraction and Trapping Techniques
The efficiency of headspace analysis can be influenced by the choice of complementary techniques. Research on food flavorings compared several volatile trapping methods and found that Stir Bar Sorptive Extraction (SBSE) detected the highest number of volatiles, proving particularly comprehensive for components like polysulfides, pyrazines, and terpene alcohols [49]. In contrast, Solid-Phase Microextraction (SPME) was more suitable for sesquiterpenes, and Dynamic Headspace (DHS) excelled at extracting monoterpenes [49]. This highlights that the choice of trapping or extraction technique within a headspace method must be aligned with the specific target analytes.
Furthermore, modern techniques like Accelerated Solvent Extraction (ASE) have been shown to outperform traditional methods like Soxhlet extraction in terms of speed, solvent consumption, and environmental impact, while delivering comparable analytical results [50]. Such advancements in sample preparation can integrate effectively with headspace GC analysis.
Essential Research Reagents and Materials
A robust headspace GC analysis relies on several key consumables and reagents. The following table details essential items and their functions.
Table 3: Research Reagent Solutions for Headspace GC Analysis
| Item | Function / Application |
|---|---|
| Headspace Vials | Gas-tight vials (e.g., 20 mL) for containing the sample and maintaining headspace pressure during incubation [47]. |
| Inlet Septa | Septa create a seal in the GC inlet. Performance varies; some modern septa (e.g., Merlin Microseal) offer longer lifetimes and reduce analyte degradation compared to standard silicone rubber septa [51]. |
| GC Columns | The stationary phase for analyte separation. For residual solvents, mid-polarity columns like the VF-624ms are commonly used [47]. |
| Internal Standards | Deuterated or structurally similar compounds used to correct for sample-to-sample variation and improve quantitative accuracy. |
| Spiking Solutions | Certified reference standard solutions of target solvents used for method validation and recovery experiments, as in the spike recovery tests for drug products [47] [52]. |
| Zero Air | High-purity air, free of volatiles, used as a make-up gas in SIFT-MS and for flushing syringes to prevent carryover [47]. |
Detailed Experimental Protocols
Protocol: Static Headspace-GC for Pharmaceutical Residual Solvents
This protocol is adapted from a comparative study of GC-FID and SIFT-MS, which followed the principles of USP <467> [47].
1. Sample Preparation:
- Prepare standard solutions and samples in an appropriate solvent, such as water or N,N-dimethylacetamide (DMAC), depending on the solubility of the drug product [47].
- For accuracy and recovery tests, spike the drug product (e.g., acetaminophen tablets or oral suspension) with known concentrations of the target residual solvents.
2. Instrumental Configuration and Conditions:
- Autosampler: GERSTEL MPS Robotic Pro or equivalent.
- Headspace Conditions:
- GC-FID Conditions (following USP <467>):
3. Data Acquisition and Analysis:
- Acquire chromatograms and integrate peak areas.
- Construct a calibration curve using the standard solutions.
- Calculate the concentration in samples using the calibration curve. For recovery, compare the measured amount in the spiked sample to the known added amount.
Protocol: Dynamic Headspace (Purge and Trap) for Trace Volatiles
This protocol outlines a generic dynamic headspace method, applicable to samples like water or solids.
1. Sample Preparation:
- Place a measured volume or mass of sample into a dedicated purge-and-trap vessel.
- Add internal standards if required.
2. Instrumental Configuration and Conditions:
- Purge Stage:
- Purge Gas: High-purity Helium or Nitrogen.
- Purge Flow Rate: ~20-40 mL/min.
- Purge Time: ~10-15 minutes.
- Sample Temperature: Controlled (e.g., ambient to 40°C).
- Trap Configuration:
- The trap typically contains a series of adsorbents (e.g., Tenax, silica gel, charcoal) to retain a broad range of volatiles.
- The trap is held at ambient or slightly sub-ambient temperature during purging to ensure efficient trapping.
- Desorb Stage:
- Desorb Temperature: Rapid heating of the trap to ~180-250°C.
- Desorb Time: ~2-4 minutes.
- The trap is backflushed with carrier gas to transfer the desorbed analytes to the GC column.
3. GC-MS Analysis:
- The desorbed analytes are separated on a GC column and detected by a Mass Spectrometer (MS).
- Analytes are quantified against calibration standards processed using the same purge-and-trap method.
Both static and dynamic headspace GC are powerful techniques for solvent analysis, yet they serve different primary purposes. Static headspace is ideal for relatively simple matrices and high-throughput analyses where the target solvents are sufficiently volatile, such as in pharmaceutical quality control labs following USP guidelines [47] [12]. Its simplicity and robustness are its greatest strengths. In contrast, dynamic headspace provides superior sensitivity and is the method of choice for trace-level analysis of volatile organic compounds in complex matrices like environmental samples (water, soil, and air) [12].
The choice between them should be guided by the analytical requirements: static headspace for routine, high-throughput applications, and dynamic headspace for challenging, low-concentration analytes. Furthermore, as instrumental technology advances, techniques like SIFT-MS demonstrate that moving beyond chromatography can dramatically increase throughput while maintaining data quality, offering a compelling alternative for specific application scenarios [47].
Static headspace gas chromatography (SHS-GC) is a widely used technique for the analysis of volatile organic compounds, yet analysts frequently encounter significant challenges with complex matrices. Common issues include poor sensitivity for low-concentration analytes, difficulties with solid or semi-solid samples, and inefficient extraction of polar or less volatile compounds that have high distribution constants and prefer to remain in the sample matrix rather than partition into the headspace [14]. These limitations are particularly problematic in pharmaceutical development and food safety applications where precise quantification of trace-level volatiles is essential for quality control and regulatory compliance [53] [54].
Two advanced dynamic headspace techniques have emerged to address these challenges: the Full Evaporative Technique (FET) and the Multi-Volatiles Method (MVM). Both methods represent significant advancements in headspace sampling technology, offering enhanced sensitivity, greater comprehensiveness, and improved capability for dealing with challenging matrices that traditionally frustrate conventional static headspace approaches [14]. This guide provides a systematic comparison of these advanced techniques against traditional static and dynamic headspace methods, supported by experimental data and detailed protocols to inform researchers and method development scientists in their selection of appropriate analytical approaches.
Technical Foundations: FET and MVM Explained
Full Evaporative Technique (FET)
The Full Evaporative Technique (FET) is a dynamic headspace sampling approach that utilizes a relatively small sample volume (typically <100 μL) in a standard 10- or 20-mL headspace vial, where both the sample and matrix are fully evaporated [14]. This technique is particularly valuable when analyzing volatile and semi-volatile compounds with high distribution constants that would otherwise prefer to remain within the sample matrix or diluent rather than volatilize into the headspace. This situation commonly occurs with polar analytes in aqueous matrices or compounds with low vapor pressure, even under elevated extraction temperatures or increased agitation [14].
The fundamental principle of FET involves complete evaporation of the sample, which effectively eliminates the matrix effect by removing the condensed phase. This approach significantly improves the partitioning of less volatile analytes into the headspace, thereby enhancing sensitivity for compounds that are challenging to analyze using conventional static headspace techniques. Research demonstrates that FET exhibits a notable bias toward compounds with higher distribution constants, showing significantly improved response for analytes at later elution times, which typically represent higher boiling or more polar compounds [14].
Multi-Volatiles Method (MVM)
The Multi-Volatiles Method (MVM) represents a more comprehensive approach to headspace analysis, designed to ensure that all volatile compounds of interest are identified and quantified, even within highly complex matrices [14]. This technique employs the full evaporation technique with sequential dynamic headspace extractions under different temperature and flow conditions, utilizing different thermal desorption trap materials to capture analytes with a wide range of chemical properties and distribution constants.
The MVM approach can be implemented in two primary configurations: (1) analyzing each thermal desorption tube separately to "fractionate" the headspace components, or (2) when combined with a cryo-inlet, desorbing each tube into the inlet and introducing the total output onto the GC column to obtain a "full profile" analysis [14]. Typically, most applications utilize three different packing varieties for the desorption tubes, selected to match the specific analyte chemistries associated with each fraction. This comprehensive extraction technique is significantly more powerful than static headspace GC for untargeted aroma profiling and complex mixture analysis [55].
Comparative Experimental Data: Performance Metrics
Sensitivity and Detection Limits
Table 1: Detection Limit Comparison Between Static and Dynamic Headspace Techniques
| Analytical Technique | Detection Limits | Matrix | Key Advantage |
|---|---|---|---|
| Static Headspace [33] | 10 ppb | Aqueous standard | Suitable for prescreening |
| Dynamic Headspace (DHS) [33] | 0.5 ppb | Aqueous standard | 20-60x lower detection limits than SHS |
| FET-DHS [14] | Significant improvement for high distribution constant analytes | Herbal-based liquor | Enhanced sensitivity for less volatile compounds |
| SHS-GC-MS [53] | <1 ppm | β-cyclodextrin | Sufficient for regulatory compliance |
Table 2: Quantitative Performance Characteristics
| Technique | Linear Range | R² | Recovery Rates | RSD |
|---|---|---|---|---|
| SHS-GC-MS [53] | 0.05-10 mg/L | >0.99 | 91.7-106.0% | 1.0-8.9% |
| FET-MVM [14] | Extended dynamic range | Not specified | Comprehensive extraction | Automated operation |
Extraction Efficiency and Comprehensiveness
Comparative studies demonstrate the superior extraction capability of dynamic headspace techniques, particularly for complex matrices. In an analysis of dry tea samples, dynamic headspace sampling proved more comprehensive and sensitive than static headspace, while also effectively handling solid samples (1g of dry tea) without additional preparation [14]. The FET approach specifically enhances the response for compounds with higher distribution constants, effectively shifting the analytical bias toward higher boiling or more polar compounds that are typically challenging for conventional static headspace [14].
When comparing headspace techniques for chemical screening of volatiles in complex plant matrices like Myrtus communis L., dynamic approaches such as headspace solid-phase microextraction (HS-SPME) have demonstrated superior performance for collecting more volatile compounds compared to classical techniques like hydrodistillation [56]. The MVM approach further extends this capability by providing a comprehensive extraction technique that captures a wider volatility range and diverse chemical functionalities in a single analytical run [14] [55].
Experimental Protocols and Methodologies
FET-DHS Protocol for Trace-Level Analytes in Herbal-Based Liquor
Sample Preparation:
- Introduce a small sample volume (<100 μL) into a 10- or 20-mL headspace vial [14]
- Ensure complete evaporation of both sample and matrix under controlled conditions
Dynamic Headspace Sampling Parameters:
- Purge the headspace continuously with inert gas (helium or nitrogen) [14] [55]
- Trap volatiles onto an appropriate adsorbent material (typically Tenax for aroma compounds) [55]
- Apply dry purge step when analyzing aqueous matrices (2500 mL helium for wine, 1500 mL for yoghurt) to remove excess water and alcohol [55]
Thermal Desorption and GC Analysis:
- Desorb trapped analytes at elevated temperature (280°C for 6 minutes) [55]
- Focus analytes on a cold trap maintained at -40°C [55]
- Rapidly heat the inlet to inject compounds onto the GC column
- For less volatile analytes, employ cryo- or cold-trapping to refocus analytes during thermal desorption [14]
MVM-GC-MS Protocol for Complex Aroma Profiling
Sample Preparation:
- Place representative sample amount (5 mL liquid, 1-2 g solid) in 20-mL headspace vials [55]
- For solid samples, consider particle size reduction to enhance extraction efficiency
Sequential Dynamic Headspace Extraction:
- Perform multiple extractions under varying temperature and flow conditions [14]
- Utilize different thermal desorption trap materials for each extraction sequence
- Select adsorbent materials based on target analyte chemistries (typically three different packing varieties) [14]
Instrumental Analysis:
- Couple with high-resolution GC-TOF-MS for untargeted analysis [55]
- Operate mass spectrometer in full scan mode with high acquisition speed for optimal deconvolution
- Consider comprehensive 2D-GC for highly complex samples
Optimization Strategies for Challenging Matrices
For solid matrices or analytes with high distribution constants, several optimization approaches can enhance extraction efficiency:
- Temperature Optimization: Systematically evaluate equilibration temperatures (typically 60-80°C for SHS [53], may be higher for FET applications)
- Salting Out: While ammonium sulfate may be more efficient than sodium chloride for polar analytes [14], note that salt addition may adversely affect recovery efficiency in some applications [53]
- Co-solvent Addition: Utilize higher boiling point diluents and other additives that promote analyte partitioning into the headspace, particularly for non-polar matrices [14]
- Automated Method Development: Leverage the automation capabilities of modern DHS systems to conduct unattended optimization campaigns using design of experiments (DoE) approaches [14]
Analytical Workflow Visualization
Figure 1: FET-DHS Analytical Workflow. The process begins with minimal sample volume followed by complete evaporation, dynamic extraction, and focused injection for enhanced sensitivity [14] [55].
Figure 2: MVM Sequential Extraction Workflow. Multiple dynamic extractions under varying conditions capture diverse volatile compounds, with options for fractionated or comprehensive analysis [14].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Advanced Headspace Analysis
| Item | Function/Purpose | Application Notes |
|---|---|---|
| Tenax TA Traps [55] | Adsorbent for volatile trapping | High retention of aromas, low water/ethanol affinity |
| Multiple Adsorbent Tubes [14] | Fractionation of different chemical classes | Typically 3 packing varieties for MVM |
| Headspace Grade Solvents [54] | Sample dissolution with minimal interference | DMSO, DMF, DMAC for water-insoluble samples |
| Ammonium Sulfate [14] | Salting out agent for polar analytes | More efficient than NaCl for some applications |
| Inert Purge Gases [14] [55] | Dynamic headspace extraction | Helium or nitrogen, high purity |
| Internal Standards [53] [56] | Quantification and method validation | Toluene-d8 for SHS, menthol for SDME |
Application Case Studies
Food Safety: Residual Solvents in β-Cyclodextrin
In food additive safety monitoring, static headspace GC-MS has been successfully optimized for detecting residual trichloroethylene (TCE) and toluene (TOL) in β-cyclodextrin. The optimized method used 100 μL injection volume, equilibrium conditions of 60°C for 45 minutes, and notably excluded salt addition which adversely affected recovery efficiency. This approach achieved coefficients of determination (R²) greater than 0.99 for both analytes across 0.05-10 mg/L concentrations, with recovery rates of 91.7-106.0% and RSDs of 1.0-8.9% [53]. Both TCE and TOL were detected below the 1 ppm regulatory limit in commercial β-CD samples, demonstrating the method's suitability for food safety applications [53].
Flavor and Fragrance Analysis
Dynamic headspace sampling has shown exceptional performance in untargeted aroma profiling of complex food matrices such as strawberry yoghurt, chocolate, and red wine [55]. When coupled with GC-TOF-MS, DHS provides exhaustive extraction covering a wide volatility range, enabling the detection of key aroma compounds and potential off-flavors. The technique is particularly valuable for characterizing aromas in challenging matrices like chocolate, where DHS is performed at 80°C with a 1000 mL sampling volume without dry purge requirements [55]. This application demonstrates the technique's versatility across diverse sample types and its capability to handle matrices with varying water and fat content.
Pharmaceutical Testing: Organic Volatile Impurities
The analysis of organic volatile impurities (OVIs) in pharmaceuticals requires careful solvent selection to achieve appropriate sensitivity while avoiding interference. Headspace-grade solvents such as DMSO are essential for dissolving water-insoluble samples while maintaining low background interference [54]. The selection of appropriate solvents is critical, as the分配系数 (K) influences analyte partitioning into the headspace, with lower values providing higher sensitivity [54]. This application highlights the importance of matrix considerations in method development and the need for high-purity reagents to avoid chromatographic interference.
The selection of headspace sampling technique should be guided by analytical requirements, matrix characteristics, and target analyte properties. Static headspace remains suitable for relatively simple matrices and high-concentration volatiles where minimal sample preparation is desired [53]. Conventional dynamic headspace techniques provide significantly enhanced sensitivity (20-60× lower detection limits) and are preferable for trace-level analysis [33]. The Full Evaporative Technique offers distinct advantages for challenging applications involving polar analytes, less volatile compounds, or matrices where distribution constants limit conventional headspace sensitivity [14]. For the most comprehensive analysis of complex volatile profiles, the Multi-Volatiles Method represents the state-of-the-art, enabling complete characterization of samples containing analytes with widely varying chemical properties [14] [55].
While FET and MVM approaches involve greater instrumental complexity and more variables to optimize compared to static headspace, their enhanced sensitivity, comprehensive extraction capabilities, and ability to handle challenging matrices make them invaluable tools for advanced analytical applications in pharmaceutical development, food safety, and flavor research. The implementation of these techniques is further facilitated by modern automation capabilities that allow unattended method optimization and operation, making these powerful approaches increasingly accessible to analytical laboratories.
Optimizing Performance: Troubleshooting Common Challenges in Headspace Analysis
In the analysis of volatile organic compounds, particularly for residual solvents in pharmaceuticals, headspace gas chromatography (HS-GC) serves as a premier technique for sample introduction. The broader thesis of comparing static and dynamic headspace reveals a fundamental trade-off: static headspace (SHS) operates as an equilibrium technique prized for its simplicity and robustness, while dynamic headspace (DHS), or purge and trap, is an exhaustive extraction method capable of achieving lower detection limits [4] [57]. For researchers and drug development professionals, the choice between these techniques hinges on the required sensitivity and the nature of the sample matrix. This guide focuses on the optimization of SHS, a method where the sample is incubated in a sealed vial until the volatile analytes reach equilibrium between the sample and the gas phases [58]. The subsequent analysis of this gas phase—the headspace—minimizes the introduction of non-volatile matrix components into the GC system, leading to cleaner samples, reduced instrument maintenance, and higher uptime [58] [57].
The fundamental goal of SHS optimization is to maximize the concentration of the target analytes in the headspace to enhance detector response. This is governed by the principle expressed in the equation A ∝ CG = C0/(K + β), where the detector response (A) is proportional to the gas phase concentration (CG), which is determined by the initial analyte concentration (C0), the partition coefficient (K), and the phase ratio (β) [58] [10]. The parameters of equilibration time, temperature, and vial pressure directly influence this equation, determining the method's sensitivity, precision, and overall success. The following sections will dissect each of these key parameters, providing structured experimental data and protocols to guide method development.
Before a detailed parameter examination, understanding the core distinctions between static and dynamic headspace is crucial. The following table provides a high-level comparison of these two principal techniques.
Table 1: Fundamental Comparison of Static and Dynamic Headspace Techniques
| Feature | Static Headspace (SHS) | Dynamic Headspace (DHS/Purge & Trap) |
|---|---|---|
| Basic Principle | Equilibrium sampling of the vapor phase above a sample [10]. | Continuous extraction and trapping of volatiles from the sample [4] [57]. |
| Process | Sample is heated in a closed vial; an aliquot of the equilibrated headspace is injected [58]. | Inert gas purges the sample, volatiles are trapped on a sorbent, then thermally desorbed into the GC [57]. |
| Key Strength | Simplicity, robustness, minimal maintenance, compatibility with complex matrices [58] [4]. | Higher sensitivity and lower detection limits due to exhaustive analyte extraction [4] [57]. |
| Typical Detection Limits | Parts-per-billion (ppb) to low percentage levels [57]. | Lower than conventional static headspace; suitable for ultratrace analysis [10]. |
| Best Suited For | Routine analysis of volatiles in samples where analytes are in high ppb or higher (e.g., residual solvents, blood alcohol) [58] [10]. | Ultrace analysis of very low-concentration volatiles (e.g., environmental contaminants in water) [10] [57]. |
The decision to use SHS is often driven by the need for a straightforward, reliable method for relatively volatile target compounds. The optimization workflow for a static headspace method logically progresses through its most critical parameters, as visualized below.
In-Depth Parameter Analysis and Experimental Protocols
Equilibration Time
Fundamentals: Equilibration time is the duration required for the analytes to establish a stable equilibrium distribution between the sample matrix and the gas phase in the headspace vial [58]. Insufficient time leads to poor reproducibility and reduced sensitivity, as the headspace concentration is not stable.
Experimental Protocol for Determination: A robust approach to determine the minimum required equilibration time is to analyze a standard or sample at multiple time intervals [58].
- Preparation: Prepare multiple vials of a standard solution containing the target analytes at a known concentration.
- Incubation: Place the vials in the headspace sampler and incubate them at a fixed, moderate temperature.
- Sequential Analysis: Analyze the vials using a series of increasing equilibration times (e.g., 5, 10, 15, 20, 30 minutes).
- Data Analysis: Plot the peak area (or height) of each analyte against the equilibration time. The minimum equilibration time is the point where the peak response plateaus, indicating that equilibrium has been reached.
Supporting Experimental Data: A study optimizing HS-GC-MS for citrus leaf volatiles found that a 15-minute incubation at 100 °C was sufficient for their system, providing a rapid and reliable extraction [59]. In pharmaceutical residual solvent analysis, a generic method using dimethylsulfoxide (DMSO) as a diluent demonstrated that equilibration could be efficiently achieved in 10 minutes at 140 °C [60]. It is critical to note that equilibration time is sample-dependent and must be determined experimentally for each new sample type [58].
Equilibration Temperature
Fundamentals: Temperature is arguably the most critical parameter in SHS optimization. Increasing the temperature decreases the partition coefficient (K), favoring the transfer of analytes from the sample phase into the headspace, thereby increasing the detector signal [58] [10]. However, this effect must be balanced against the risk of solvent vaporization or analyte degradation.
Experimental Protocol for Optimization:
- Preparation: Prepare replicate samples of a standard.
- Temperature Gradient: Incubate the samples at a range of temperatures, typically in 10-20 °C increments. A common starting range is 40-120 °C, depending on the solvent boiling point.
- Upper Limit: A key rule is to keep the maximum oven temperature at least 20 °C below the boiling point of the sample solvent to prevent excessive pressure and vaporization [58] [26].
- Data Analysis: Plot the peak area of each analyte against temperature. The optimal temperature is the highest point that provides maximum response without causing decomposition or excessive solvent background.
Supporting Experimental Data: The profound effect of temperature is demonstrated in an experiment where a sample was equilibrated for 20 minutes across a temperature range. The chromatograms showed a clear and significant increase in detector response at higher temperatures [58]. For instance, the partition coefficient for ethanol in water decreases from ~1350 at 40 °C to ~330 at 80 °C, directly translating to more analyte in the headspace [58]. The study on kimchi analysis further highlights this, where the cool injection system (CIS) temperature was optimized to -40 °C to achieve the highest trapping efficiency for volatile compounds [61].
Table 2: Summary of Temperature Optimization Findings from Literature
| Application Context | Optimal Temperature Finding | Key Experimental Observation |
|---|---|---|
| Generic Pharmaceutical Solvents [60] | 140 °C with DMSO diluent | Enabled efficient 10-min equilibration; DMSO's high boiling point (189°C) allowed high-temperature operation. |
| Theoretical Guidance [58] [26] | 20 °C below solvent boiling point | Critical rule to prevent excessive vial pressure and solvent vaporization. |
| Kimchi Volatiles (HS-HIT) [61] | CIS at -40 °C | Optimized trapping in a cooled injection system, maximizing sensitivity. |
Vial Pressure
Fundamentals: In automated valve-and-loop SHS systems, the vial is pressurized with carrier gas before sampling. The primary role of pressure is mechanical, not thermodynamic. According to equilibrium principles, adding an inert gas at constant volume does not shift the chemical equilibrium or change the partition coefficient [10]. Instead, higher vial pressure ensures consistent and rapid transfer of the headspace aliquot into the sample loop and transfer line.
Experimental Protocol for Optimization:
- Parameter Identification: The key parameters to optimize are the pressurization pressure and the pressurization time.
- Testing: Analyze replicate standard samples at different pressurization pressures (e.g., 5, 10, 15 psi) while keeping all other parameters constant.
- Evaluation: Assess the impact on peak area reproducibility (precision) and sensitivity. The goal is to find the minimum pressure and time required for a stable and reproducible injection without affecting the analyte mixture.
Supporting Experimental Data: While pressure has a minor influence on the partition coefficient, it is crucial for the mechanics of the injection. Sufficient pressurization is essential for achieving consistent and reliable results [26]. The pressurization step is part of the standard three-step process in modern headspace samplers: 1) pressurize the vial, 2) vent to fill the loop, and 3) inject the contents onto the GC column [58]. Modern instruments have tools to help determine optimal pressures for a given sample type [58].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful and reproducible SHS analysis depends on the selection of appropriate consumables and reagents. The following table details key items and their functions.
Table 3: Essential Materials for Static Headspace Analysis
| Item | Function & Importance |
|---|---|
| High-BoPoint Diluent (e.g., DMSO) | Dissolves drug substances and allows for high equilibration temperatures without vaporization, increasing sensitivity and reducing equilibration time [60]. |
| Headspace Vials | Vials must be sealed to prevent loss of volatiles. Larger vials (10-22 mL) allow for a larger sample volume and/or headspace, which can be optimized via the phase ratio (β) [58]. |
| Septum & Caps | Must form a tight seal and withstand the vial equilibration temperature without degrading or leaking [58] [26]. |
| Narrow Bore GC Inlet Liner | Prevents band broadening during the injection of the gaseous sample, leading to sharper peaks and better sensitivity [26]. |
| Non-Volatile Salts (e.g., NaCl) | Induces the "salting-out" effect in aqueous samples, reducing the solubility of analytes and driving them into the headspace, thereby increasing their concentration [26]. |
The optimization of static headspace for solvents research is a systematic process centered on three interdependent parameters: equilibration time, temperature, and vial pressure. As detailed in this guide, temperature is the most powerful lever for increasing sensitivity by driving analytes into the headspace, while equilibration time must be sufficient to ensure a stable, reproducible equilibrium state. Finally, vial pressure is a key practical parameter for ensuring consistent instrument operation and injection precision. A methodical approach to optimizing these factors, grounded in the fundamental principle of phase equilibrium, will yield a robust, sensitive, and reliable SHS-GC method suitable for the demanding requirements of pharmaceutical research and drug development.
In the context of solvents research, particularly for pharmaceutical and food safety, the analysis of residual solvents and volatile organic compounds (VOCs) is a critical requirement. While static headspace (SHS) techniques are widely used for their simplicity, dynamic headspace (DHS) sampling, also known as purge and trap, has emerged as a powerful alternative that addresses several SHS limitations. DHS provides enhanced sensitivity and a more comprehensive extraction of volatile analytes, especially for compounds with high distribution constants that prefer to remain in the sample matrix rather than partition into the headspace [36] [14]. This technique involves continuously purging the headspace of a sample with an inert gas, transferring the volatiles onto a sorbent trap, followed by thermal desorption into a gas chromatography (GC) system.
The primary advantage of DHS lies in its ability to overcome the equilibrium limitations of SHS. By continuously removing analytes from the headspace, the system promotes further release of volatile compounds from the sample matrix, leading to significantly improved extraction efficiency, particularly for trace-level analytes and those challenging polar or semi-volatile compounds [36] [14]. However, achieving optimal performance requires meticulous optimization of several interdependent parameters, chief among them being purge flow rate, sorbent trap selection, and desorption conditions. This guide provides an objective comparison of these key parameters and their optimization, supported by experimental data from recent research.
Comparative Analysis: Static Headspace vs. Dynamic Headspace
Before delving into parameter optimization, it is essential to understand the fundamental operational differences and performance characteristics of static and dynamic headspace techniques, as this forms the core thesis of modern solvent analysis method selection.
Table 1: Comparison of Static Headspace (SHS) and Dynamic Headspace (DHS) Techniques
| Feature | Static Headspace (SHS) | Dynamic Headspace (DHS) |
|---|---|---|
| Basic Principle | Equilibrium-based sampling of the vapor phase in a closed system [4]. | Continuous purging of the headspace, transferring analytes to a sorbent trap [4]. |
| Sensitivity | Limited by equilibrium concentration; generally lower sensitivity [14]. | Superior sensitivity due to exhaustive extraction and analyte preconcentration on the trap [62] [63]. |
| Suitability for Trace Analysis | Less ideal for very low concentration analytes [14]. | Excellent for trace-level analysis; can achieve signal intensities up to 450 times greater than SHS and HS-ITEX [62]. |
| Handling of Challenging Matrices | Struggles with solid matrices, polar analytes in polar matrices, and less volatile compounds [14]. | Effectively handles solid samples, aqueous matrices (with dry purge), and a wider volatility range [36] [14]. |
| Key Optimization Parameters | Equilibration temperature and time, sample-to-headspace volume ratio, salting-out [24]. | Purge flow rate and volume, sorbent trap selection, desorption temperature and time [36] [63]. |
| Throughput & Simplicity | Simple, fast, and easily automated [4]. | More complex setup and longer method development, but fully automatable [36]. |
| Risk of Artifacts | Lower risk as no sorbent is used. | Potential for artifact formation or analyte breakdown on the sorbent trap if improperly selected [4]. |
Core DHS Parameters and Optimization Strategies
The performance of a DHS method is governed by a triad of critical parameters. Their optimization is not independent, and a Design of Experiments (DoE) approach is highly recommended to understand interaction effects [36] [64].
Purge Flow Rate and Volume
The purge flow rate and the total purge volume are fundamental parameters that control the efficiency of transferring analytes from the headspace to the sorbent trap.
- Function: The purge gas stream strips volatile compounds from the headspace above the sample. An optimal flow rate must be high enough to efficiently transfer analytes but not so high that it causes breakthrough on the trap (where compounds are not retained) or creates excessive water vapor from aqueous samples [36].
- Experimental Optimization: In a study optimizing furfurals in vinegar, a Full Factorial Design (FFD) was employed, identifying purge volume as a significant factor. The optimized method used a purge volume of 800 mL [63]. Another study on food volatiles used a Box-Behnken experimental design to simultaneously optimize purge flow rate and volume, finding that these parameters significantly impacted the total summed peak area and number of detected compounds [36].
Sorbent Trap Selection
The sorbent trap is the heart of the DHS system, responsible for concentrating the analytes. The choice of sorbent material directly determines the spectrum of compounds that can be effectively captured and analyzed.
- Function: Sorbent traps contain one or more materials that adsorb volatile compounds based on their chemical properties. Their higher capacity compared to SPME fibers allows for the extraction of larger quantities of analytes [63].
- Common Sorbents and Applications:
- Tenax TA: A hydrophobic polymer (polyphenylene oxide) excellent for trapping a wide range of VOCs (C7-C30) and is highly resistant to water. It was successfully used for trapping VOCs from sourdough and furfurals from vinegar [36] [63].
- Tenax GR: A composite of Tenax TA and graphite, offering similar properties with altered selectivity for certain compounds [63].
- Multi-bed Traps: Traps containing multiple sorbents (e.g., Carbopack X, Carboxen) are available for a very broad range of analytes, from highly volatile to semi-volatile compounds [14].
- Experimental Data: The performance of Tenax TA and Tenax GR was directly compared for furfural analysis, with Tenax TA being selected for the optimized method [63]. The life of a well-maintained DHS trap can be significantly longer, up to 10 times, than an SPME fibre [62].
Desorption Conditions
Thermal desorption is the process of transferring trapped analytes from the sorbent tube into the GC inlet. Its efficiency is critical for achieving good sensitivity and peak shape.
- Function: The sorbent trap is rapidly heated in a reverse flow of carrier gas to release the concentrated analytes, which are then transferred to the GC column. Incomplete desorption can lead to carryover and memory effects between runs [36].
- Key Parameters: Desorption temperature and desorption time are the two most critical factors. The temperature must be high enough to rapidly release all target analytes from the sorbent. The hold time must be long enough to ensure complete transfer.
- Experimental Optimization: For the analysis of furfurals, the TDU hold time was optimized via a FFD, with a 5-minute desorption time being part of the final optimized conditions [63]. For sourdough VOC analysis, a desorption temperature program up to 250 °C was used to ensure complete release of all trapped compounds [36].
Table 2: Summary of Optimized DHS Parameters from Cited Experimental Studies
| Study Focus | Optimized Purge Volume | Optimized Sorbent | Optimized Desorption Conditions | Analytical Technique |
|---|---|---|---|---|
| Furfurals in Vinegar [63] | 800 mL | Tenax TA | TDU hold time: 5 min | GC/MS |
| VOCs in Sourdough [36] | Determined via Box-Behnken DoE | Tenax TA | Initial temp: 50°C, ramped to 250°C at 720°C/min, held 10 min | GC×GC–TOF-MS |
| Pharmaceutical Residual Solvents [64] | Not Specified | Not Specified | Oven IT: 30°C, FT: 158°C (after inlet desorption) | HSGC-FID |
Experimental Protocols for DHS Optimization
Generalized Workflow for DHS Method Development
The following diagram illustrates a logical, step-by-step workflow for developing and optimizing a Dynamic Headspace method, incorporating the key parameters discussed.
Detailed Methodology from a Vinegar Analysis Study
The following protocol is adapted from a study that optimized a DHS procedure for determining furfurals in vinegar using a Full Factorial Design (FFD) [63].
- Sample Preparation: An "artificial matrix" mimicking the physical/chemical characteristics of Aceto Balsamico Tradizionale di Modena (ABTM) was used for quantification due to the lack of certified reference materials.
- DHS Instrumental Parameters:
- Incubation Temperature: 40 °C (optimized).
- Incubation Time: 10 min (optimized).
- Purge Volume: 800 mL (optimized).
- Dry Purge Volume: 1500 mL (to remove water vapor).
- Sorbent: Tenax TA.
- Thermal Desorption:
- TDU Hold Time: 5 min (optimized).
- GC-MS Analysis: Trapped analytes were thermally desorbed and analyzed by GC-MS.
- Optimization Design: A FFD was used to evaluate the effects of five factors (incubation temperature, incubation time, purge volume, dry volume, and TDU hold time). Center points and their replicates were added to check for non-linear relationships and evaluate method precision. Data was evaluated using analysis of variance (ANOVA) and Principal Component Analysis (PCA).
The Scientist's Toolkit: Essential Reagents and Materials
Successful DHS analysis relies on a set of key reagents and materials. The following table details these essential components and their functions.
Table 3: Key Research Reagent Solutions and Materials for DHS
| Item | Function / Application | Examples / Specifications |
|---|---|---|
| Sorbent Tubes | Adsorption and concentration of volatile analytes from the purge gas stream. | Tenax TA (for hydrocarbons, VOCs); Multi-bed traps (e.g., Carbotrap, Carboxen) for a wider volatility range [36] [63]. |
| High-Purity Purge Gas | Inert gas used to purge volatiles from the sample headspace. | Nitrogen or Helium, high-purity grade (e.g., 4.8 or 5.0) to avoid introducing contaminants [36]. |
| Headspace Grade Solvents | High-purity solvents for dissolving samples to prevent interference from volatile impurities. | DMSO, DMF, DMAC; specifically tested for low volatile background [65]. |
| Internal Standards | Compounds added to sample to correct for losses and variability during sample prep and injection. | Should be chemically similar, well-resolved from analytes, and not present in the sample [64]. |
| Design of Experiments Software | Statistical software for efficient experimental design and data analysis to optimize multiple parameters. | Used to build models (e.g., Box-Behnken) and perform Response Surface Methodology [36] [64]. |
Advanced DHS Techniques
To overcome the limitations of standard DHS with complex matrices, several advanced techniques have been developed.
- Full Evaporative Technique (FET): This technique uses a very small sample volume (typically <100 µL) in a standard headspace vial, allowing the sample and matrix to be fully evaporated. This is particularly useful for analytes with high distribution constants or for samples where the matrix is problematic (e.g., viscous or solid). FET-DHS has been shown to provide a significant sensitivity boost for less volatile and polar compounds compared to conventional DHS [14].
- Multi-Volatiles Method (MVM): This comprehensive approach involves sequential dynamic headspace extractions under different temperature and flow conditions, and sometimes using different sorbent trap materials. The aim is to fractionate the headspace to capture a very wide range of analytes with differing chemistries and volatilities. Each trap can be analyzed separately or desorbed into a single cryo-inlet for a full volatile profile [14].
The optimization of purge flow dynamics, sorbent trap chemistry, and thermal desorption parameters is paramount to unlocking the full potential of Dynamic Headspace analysis. While the method development process is more complex than for Static Headspace, the payoff is substantially higher sensitivity and the ability to analyze a much broader range of compounds and matrices. The experimental data and protocols summarized herein demonstrate that a systematic, DoE-driven approach is the most effective path to a robust and reliable DHS method. For researchers in pharmaceutical development and food safety requiring the utmost sensitivity and comprehensive volatile profiling, DHS and its advanced variants like FET and MVM present a powerful and often necessary analytical solution.
In the analysis of volatile and semi-volatile compounds, matrix effects present a significant challenge that can compromise the accuracy, sensitivity, and reproducibility of results. These effects occur when components of the sample matrix interfere with the extraction, separation, or detection of target analytes, leading to suppressed or enhanced signals, altered partition coefficients, and ultimately, inaccurate quantification. Within the context of static and dynamic headspace analysis for solvents research, understanding and mitigating matrix effects is paramount for method development and validation.
The fundamental principle of headspace gas chromatography (HS-GC) relies on the equilibrium partitioning of analytes between the sample matrix and the vapor phase above it. In static headspace, the system reaches equilibrium in a closed vial before a portion of the vapor is sampled, making it susceptible to matrix-induced variations in partitioning behavior [21]. In contrast, dynamic headspace continuously purges volatiles from the sample onto an adsorbent trap, which can overcome some equilibrium limitations but introduces other matrix considerations, particularly regarding trapping efficiency and desorption dynamics [43] [66]. Both techniques must contend with matrices that can range from simple aqueous solutions to complex biological, environmental, or pharmaceutical samples containing proteins, lipids, carbohydrates, and inorganic salts that potentially interact with target analytes.
This guide objectively compares strategic approaches to overcome these challenges, focusing on three powerful tools at the analyst's disposal: salting-out effects, solvent selection, and pH adjustment. By examining experimental data and optimized protocols, we provide a framework for researchers to select the most appropriate techniques for their specific analytical challenges in pharmaceutical and chemical development.
Static vs. Dynamic Headspace: A Fundamental Comparison
The choice between static and dynamic headspace sampling is foundational to how matrix effects will manifest and must be addressed. Each technique operates on different physical principles and offers distinct advantages and limitations for handling complex matrices.
Static headspace is an equilibrium-based technique where the sample is heated in a sealed vial until the volatile compounds partition between the sample matrix and the headspace vapor. At equilibrium, a portion of this vapor is injected into the GC system [21]. This technique excels in simplicity and is ideal for clean matrices and volatile analytes with favorable partition coefficients. However, its primary limitation lies in its sensitivity to the partition coefficient (K), defined as K = CS/CG, where CS and CG are the concentrations in the sample and gas phases, respectively [21]. For analytes with high solubility in the sample matrix (where K >> β, the phase ratio), even minor temperature fluctuations can significantly impact results. For instance, with soluble compounds like ethanol in water, a temperature shift from 60°C to 62°C can increase the measured peak area by nearly 10% [21]. This heightened sensitivity makes static headspace particularly vulnerable to matrix effects that alter the partition coefficient.
Dynamic headspace (DHS), also referred to as purge-and-trap, operates on a non-equilibrium principle. An inert gas continuously purges the sample headspace or bubbles through the liquid sample, sweeping volatiles onto an adsorbent trap where they are concentrated [43] [66]. This continuous removal displaces the equilibrium, theoretically allowing for near-complete extraction of volatiles. DHS generally provides significantly enhanced sensitivity and is better suited for trace-level analysis and less volatile compounds. As evidenced in comparative studies, such as the analysis of dry tea, DHS demonstrates a more comprehensive and sensitive profile than static headspace [43]. The technique is also less susceptible to matrix-induced equilibrium shifts, though it introduces additional optimization parameters such as sorbent selection, purge flow rate, and trap conditioning [36].
Table 1: Comparison of Static and Dynamic Headspace Techniques
| Feature | Static Headspace | Dynamic Headspace |
|---|---|---|
| Principle | Equilibrium-based in closed system | Non-equilibrium, continuous purging |
| Sensitivity | Limited by equilibrium concentration | Enhanced through analyte trapping and concentration |
| Matrix Effect Susceptibility | High for soluble analytes | Lower, but sorbent selection is critical |
| Optimal Use Cases | Simple matrices, highly volatile analytes, regulatory methods (e.g., USP <467>) | Complex matrices, trace analysis, broad volatility range |
| Key Optimization Parameters | Temperature, equilibration time, phase ratio (β) | Purge flow/volume, sorbent type, desorption parameters |
Strategic Approach 1: Salting-Out
Mechanism and Principles
The salting-out effect is a powerful strategy to mitigate aqueous matrix effects by manipulating the ionic strength of the solution. At its core, this technique involves adding a soluble salt to an aqueous sample, which at high concentrations reduces the solubility of organic analytes in the aqueous phase, thereby driving them into the headspace vapor or a coexisting organic solvent phase [67]. This process is fundamentally explained by the Setschenow equation: log(S0/S) = Ks × I, where S0 and S are the solubilities in pure water and salt solution, respectively, Ks is the salting-out constant, and I is the ionic strength of the solution [67]. The addition of salt increases the ionic strength, effectively "ordering" water molecules around the ions and making them less available to solvate neutral organic molecules. This enhancement of the hydrophobic effect promotes the partitioning of analytes out of the aqueous matrix.
Salt Selection and the Hofmeister Series
The efficiency of salting-out depends significantly on the choice of salt. The Hofmeister series provides a valuable guide, ranking ions based on their ability to precipitate (salt out) proteins and other molecules [67]. Generally, small, multiply charged ions (kosmotropes) are more effective at salting out than larger, singly charged ions (chaotropes).
Table 2: Salt Selection Based on the Hofmeister Series for Salting-Out
| Relative Effectiveness | Anions | Cations |
|---|---|---|
| Most Effective | CO₃²⁻, SO₄²⁻, HPO₄²⁻ | Mg²⁺, Ca²⁺, Li⁺ |
| Moderately Effective | F⁻, Cl⁻ | Na⁺, K⁺ |
| Less Effective (Can Cause Salting-In) | SCN⁻, I⁻, ClO₄⁻, NO₃⁻ | NH₄⁺, Rb⁺, Cs⁺ |
In practice, magnesium sulfate (MgSO₄) and sodium chloride (NaCl) are frequently employed. MgSO₄ is particularly effective due to the high charge density of both ions. Research indicates that ammonium sulfate can provide superior extraction efficiency for polar analytes from polar matrices compared to sodium chloride, potentially allowing for lower salt quantities [43]. The QuEChERS (Quick, Easy, Cheap, Effective, Rugged, and Safe) method for pesticide analysis in produce famously utilizes a combination of MgSO₄ and sodium citrate salts to drive polar pesticides into the acetonitrile phase [67].
Experimental Protocol and Data
SALLE Protocol for Pharmaceutical Analysis (e.g., Ciprofloxacin in Water) [68]:
- Sample Preparation: Transfer 10 mL of aqueous sample (e.g., water, buffered solution) to a 15 mL centrifuge tube.
- Salt Addition: Add 4 g of anhydrous MgSO₄ to the sample. The high mass of salt ensures sufficient ionic strength to induce phase separation.
- Solvent Addition: Add 5 mL of acetonitrile (a water-miscible solvent).
- Mixing: Shake the mixture gently for 6 minutes to ensure complete dissolution of the salt and interaction between phases.
- Centrifugation: Centrifuge at 4000 rpm for 5 minutes. This will result in the clear separation of the organic phase (acetonitrile, now containing the analytes) from the salted aqueous phase.
- Analysis: Carefully withdraw the upper organic layer for direct analysis or concentrate it under a gentle stream of nitrogen and reconstitute in a compatible solvent for HPLC or GC analysis.
This SALLE method has demonstrated excellent performance for the extraction of ciprofloxacin from water samples, achieving a limit of detection (LOD) of 0.075 μg/L and recoveries ranging from 86.4% to 120% across tap, mineral, and wastewater matrices [68]. Similarly, SALLE has been successfully applied to the simultaneous determination of pyrrolizidine alkaloids (PAs) and their N-oxides (PANOs) in honey and pollen, showing excellent extraction efficiency (82–104%) and recoveries (82–113%) for most target analytes, with negligible matrix effects [69].
Diagram: Salting-Out Assisted Liquid-Liquid Extraction (SALLE) Workflow
Strategic Approach 2: Solvent Selection
Role of Solvent Polarity and Chemistry
Strategic solvent selection is critical for optimizing the partitioning of analytes and minimizing co-extraction of matrix interferents. The core principle is "like dissolves like"—matching the polarity and chemical characteristics of the solvent to those of the target analytes to maximize extraction efficiency while minimizing interference from the matrix. In the context of headspace analysis, solvents can be used as sample diluents or as the extractant phase in salting-out procedures.
The polarity of the solvent directly influences which analytes will be preferentially extracted. For reverse-phase chromatography and related extractions, common solvents are often used in combination. Acetonitrile (ACN) is a polar solvent that offers a good balance of solubility for a wide range of analytes, low viscosity, and effective protein precipitation, making it a common choice in QuEChERS and SALLE methods [67] [70]. Methanol (MeOH), also highly polar, can sometimes provide better selectivity for certain hydrophobic peptides but suffers from higher viscosity, which can lead to broader chromatographic peaks [70]. For non-polar analytes, hexane or ethyl acetate are frequently employed in normal-phase extraction protocols [70].
Solvent Selection in Headspace Techniques
In static headspace, the sample is often dissolved or diluted in a solvent. A high-boiling-point solvent is typically chosen to avoid overwhelming the headspace vapor volume and the chromatographic system. The Full Evaporative Technique (FET) represents a specialized approach where a very small sample volume (typically <100 μL) is completely evaporated in a large headspace vial (10-20 mL) [43]. This technique is particularly useful for challenging matrices like shampoos or viscous samples, as it liberates analytes that would otherwise be retained strongly by the matrix due to high distribution constants. As shown in Figure 6 of the search results, FET-DHS provided a significant improvement in the recovery of higher-boiling or more polar compounds from a shampoo matrix compared to standard DHS [43].
Table 3: Solvent Selection Guide for Different Analytical Tasks and Matrices
| Analytical Task / Matrix | Recommended Solvent System | Rationale |
|---|---|---|
| Reverse-Phase HPLC (General) | Water + Acetonitrile/Methanol | Adjustable polarity; acetonitrile offers lower viscosity for better efficiency. |
| Peptide Separation (RP-HPLC) | Water + 0.1% TFA + Acetonitrile | Acetonitrile provides sharper peaks and shorter retention times vs. methanol. |
| Normal-Phase HPLC (Lipids, Hydrocarbons) | Hexane + Ethyl Acetate | Effectively elutes non-polar to moderately polar compounds. |
| SALLE / QuEChERS | Acetonitrile (with salts) | Effective protein precipitation and partitioning for a wide polarity range of analytes. |
| Headspace Diluent (FET) | Methanol (for dilution prior to evaporation) | Aids in homogenizing the sample before full evaporation in the vial. |
Strategic Approach 3: pH Adjustment
Controlling Analyte Ionization
pH adjustment is a supremely effective strategy for manipulating the chemical form of ionizable analytes, thereby controlling their solubility and volatility. For an acid (HA ⇌ H⁺ + A⁻) or a base (BH⁺ ⇌ B + H⁺), the neutral species (HA or B) is invariably more volatile and has a higher partition coefficient into an organic solvent or the headspace vapor than its charged counterpart (A⁻ or BH⁺). The Henderson-Hasselbalch equation is used to calculate the fraction of analyte in its neutral form: for acids, pH = pKₐ + log([A⁻]/[HA]); for bases, pH = pKₐ + log([B]/[BH⁺]) [68].
The general rule is to adjust the sample pH to at least 2 units above the pKₐ for basic analytes to ensure they are in their deprotonated, neutral form, and at least 2 units below the pKₐ for acidic analytes to ensure they are in their protonated, neutral form. This suppression of ionization dramatically increases the analyte's affinity for the organic or gas phase. For example, ciprofloxacin, a fluoroquinolone antibiotic, has two pKₐ values (5.76 for the carboxylic acid group and 8.68 for the amine group) and exists as a zwitterion at neutral pH [68]. Effective extraction, therefore, requires careful pH control to manipulate its charge state and solubility.
Experimental Implementation
The implementation of pH adjustment is methodologically straightforward but requires precise execution:
- Determine pKₐ: Identify the pKₐ values of the target analytes from chemical databases or the literature.
- Select Buffer System: Choose an appropriate buffer with a pKₐ within ±1 unit of the desired pH to ensure sufficient buffering capacity. Common buffers include phosphate (pH ~2-8), acetate (pH ~3.8-5.8), and ammonium salts [70].
- Adjust pH: Prior to headspace equilibration or liquid-liquid extraction, adjust the sample pH using the selected buffer, or by careful addition of dilute acid (e.g., HCl, H₂SO₄, TFA) or base (e.g., NaOH, NH₄OH).
- Verify pH: Confirm the final pH of the solution using a calibrated pH meter.
This strategy is widely used in the analysis of pharmaceuticals, pesticides, and environmental contaminants. In the development of a SALLE method for pyrrolizidine alkaloids, pH was one of the critical parameters optimized using a chemometric approach to ensure simultaneous efficient extraction of both the free bases and their N-oxides [69].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the strategies discussed requires a well-stocked laboratory. The following table details key reagents and materials essential for methods development focused on overcoming matrix effects.
Table 4: Essential Research Reagent Solutions for Mitigating Matrix Effects
| Reagent/Material | Primary Function | Application Examples |
|---|---|---|
| MgSO₄ (Anhydrous) | Salting-out agent; creates high ionic strength to drive analytes out of aqueous phase. | QuEChERS; SALLE for pharmaceuticals and pesticides [67] [69] [68]. |
| NaCl | Salting-out agent; alternative to MgSO₄, though often less efficient. | General liquid-liquid extraction; static headspace sensitivity enhancement [67] [66]. |
| Ammonium Sulfate | Salting-out agent; can be more efficient than NaCl for polar analytes [43]. | Static headspace of polar compounds in polar matrices. |
| Acetonitrile (HPLC Grade) | Water-miscible extraction solvent; effective protein precipitant. | SALLE; QuEChERS; reverse-phase HPLC mobile phase [70] [69] [68]. |
| Methanol (HPLC Grade) | Water-miscible extraction solvent and diluent. | Reverse-phase HPLC mobile phase; sample dilution for FET [70] [43]. |
| Phosphate Buffer Salts | Provides stable pH control in aqueous solutions, crucial for ionizable analytes. | Adjusting sample pH for extraction of acidic/basic compounds [70]. |
| Acetate Buffer Salts | Provides stable pH control in the mildly acidic range. | Adjusting sample pH for extraction of acids/bases with pKₐ near 4-5 [70]. |
| Trifluoroacetic Acid (TFA) | Ion-pairing reagent and pH adjuster for acidic conditions. | Peptide and protein separations by HPLC; suppression of ionization for basic analytes [70]. |
| Tenax TA Sorbent Tubes | Traps volatiles in Dynamic Headspace; low affinity for water. | DHS-GC analysis of VOCs from complex matrices (food, polymers) [43] [36]. |
Overcoming matrix effects requires a systematic and often integrated approach. The most robust methods frequently combine salting-out, strategic solvent selection, and precise pH adjustment. The initial choice between static and dynamic headspace will dictate which strategies are most critical to optimize. For static headspace, where equilibrium is key, pH adjustment and salting-out are primary levers for manipulating the partition coefficient. In dynamic headspace, which focuses on exhaustive extraction, sorbent selection and purge conditions are paramount, though pH and solvent selection remain important for efficient analyte release from the matrix.
Modern method development increasingly relies on multivariate optimization protocols like Design of Experiments (DoE) to efficiently navigate the complex interactions between these parameters [36]. A Box-Behnken design, for instance, can simultaneously optimize incubation temperature, purge flow rate, and purge volume in DHS, leading to more robust and high-performing methods than the traditional one-factor-at-a-time approach [36].
In conclusion, matrix effects are an inevitable challenge in analytical chemistry, but they are not insurmountable. By understanding the principles of salting-out, solvent chemistry, and pH control, and by applying them within a structured method development framework, researchers and drug development professionals can reliably develop accurate, sensitive, and robust analytical methods for even the most complex sample matrices.
The analysis of low-volatility or high-affinity analytes represents a significant challenge in pharmaceutical development and environmental monitoring. These compounds, which include various residual solvents and small organic amines, often exhibit poor vapor pressure or strong matrix interactions, leading to inadequate detection limits using conventional methods. Static (SHS) and Dynamic Headspace (DHS) techniques offer divergent pathways to address these sensitivity issues, each with distinct mechanisms for enhancing analyte detection. Static Headspace involves thermostating a sample in a sealed vial to allow volatiles to reach equilibrium between the sample and the headspace, which is then sampled for injection [35]. In contrast, Dynamic Headspace techniques like In-tube Extraction (ITEX-DHS) actively purge the sample headspace and trap analytes on a sorbent material through repeated aspiring and dispensing cycles, effectively enriching analyte concentration through multiple extraction phases [71]. This comparative guide objectively evaluates both techniques' performance characteristics, supported by experimental data and detailed methodologies to inform researchers' analytical strategies for challenging compounds.
Technical Comparison: Static vs. Dynamic Headspace Mechanisms
Fundamental Operational Principles
The core distinction between SHS and DHS lies in their extraction methodologies and enrichment capabilities. SHS operates as a single-step equilibrium process where the sample is heated in a closed vial until the volatile compounds partition between the sample matrix and the headspace vapor phase. Once equilibrium is established, an aliquot of this headspace is injected directly into the GC system [35]. This method benefits from simplicity and minimal sample handling but is fundamentally limited by the equilibrium concentration of analytes in the headspace.
DHS employs a multi-stage enrichment approach where an inert gas purges the sample vessel, transferring volatile compounds from the headspace onto a specialized sorbent trap. The ITEX-DHS system achieves this through an automated process of repeated headspace extraction cycles (purge strokes), effectively concentrating analytes from a larger sample volume than SHS can access [71]. This trapped material is then thermally desorbed into the GC instrument, significantly enhancing sensitivity for trace-level analytes. Research demonstrates that "the automated ITEX Dynamic Headspace unit demonstrated equivalent accuracy and precision when compared to purge and trap and overall was more sensitive than the other enrichment techniques" while allowing for "automated analysis and optimization of conditions which drastically reduces analyst's time" [71].
Performance Metrics and Limitations
Sensitivity and Detection Limits: DHS typically provides superior sensitivity for low-concentration analytes due to its preconcentration capabilities. By continuously stripping volatiles from the sample matrix, DHS can achieve detection limits 10-1000 times lower than SHS, particularly beneficial for high-affinity compounds that resist partitioning into the headspace. The enrichment factor in DHS is directly proportional to the number of extraction cycles, allowing method customization based on sensitivity requirements [71].
Throughput and Operational Complexity: SHS offers advantages in analysis speed and operational simplicity for routine applications. With typical equilibration times of 5-15 minutes sufficient for most applications [35], SHS can support higher sample throughput for quality control environments. DHS methods, while more time-consuming per sample, provide greater sensitivity benefits that justify the additional time investment for trace analysis. Modern automated systems like ITEX-DHS have streamlined the process, reducing hands-on technician time [71].
Matrix Effects and Analyte Characteristics: SHS performance is highly dependent on the sample matrix's influence on vapor pressures and partitioning behavior. The technique shows limitations for high-boiling-point compounds (>250°C) and analytes with strong matrix interactions. DHS can mitigate some matrix effects through the purging process and is particularly effective for volatile and semi-volatile organic compounds across a broader polarity range [71]. Vacuum-assisted DHS (V-ITEX) further extends these capabilities by overcoming limitations for "polar analytes given by low vapor pressure or higher boiling points of semi-volatile compounds" through sampling at decreased pressure levels [71].
Experimental Comparison and Data Presentation
Method Parameters and Optimization Data
Experimental optimization reveals critical differences in how SHS and DHS respond to methodological adjustments. The following table summarizes key parameter effects based on amine analysis studies:
Table 1: Impact of Experimental Parameters on Static Headspace Performance
| Parameter | Optimal Range | Effect on Signal | Notes |
|---|---|---|---|
| Sample Volume | 1-2 mL | Increasing volume beyond 1 mL does not yield expected signal increases | 5 mL samples show reduced response for some amines [35] |
| Equilibration Time | 5-15 minutes | No significant increase beyond 10-15 minutes | 60-minute USP recommendation found excessive [35] |
| Diluent Composition | DMSO/Water or DMSO/NaOH mixtures | Varies significantly by analyte | Allylamine response decreased with increasing DMF [35] |
| Salt Addition | Varies by application | Can enhance vapor concentration for aqueous systems | Works best for aqueous-based solvents [35] |
Table 2: Dynamic Headspace (ITEX-DHS) Performance Characteristics
| Parameter | Configuration | Performance Advantage | Application Evidence |
|---|---|---|---|
| Extraction Mechanism | Repeated headspace aspiration through sorbent trap | Equivalent accuracy and precision to purge & trap | Suitable for heavily loaded samples without cross-contamination risk [71] |
| Automation Level | Fully automated ITEX system | Reduced analyst time vs. manual methods | Enables analysis of trace organic compounds with wide volatility range [71] |
| Sensitivity | Multiple enrichment cycles | Superior to other enrichment techniques | Effectively analyzes volatile compounds from complex matrices like honey, cheese, blood [71] |
| Matrix Tolerance | Vacuum-ITEX capability | Handles semi-volatiles with higher boiling points | Overcomes limitations of traditional DHS for polar analytes [71] |
Analytical Performance Metrics
Direct comparison studies demonstrate the contextual advantages of each technique. In pharmaceutical residual solvent analysis, SHS provides sufficient sensitivity for most USP <467> requirements with excellent precision (≤2.0% RSD typical) and minimal sample preparation [35]. For more challenging applications such as flavor compound profiling in food products or pesticide analysis in complex biological matrices, ITEX-DHS shows marked advantages, with demonstrated capability to detect "volatile organic compounds (VOCs) from honeys to differentiate plant and geographical origins" and analyze "flavors, pesticides, and biomarkers directly from complex food matrix" [71].
The sensitivity gap between techniques widens particularly for high-affinity analytes that strongly interact with their matrix. While SHS struggles to displace these compounds into the headspace, DHS continuously shifts the equilibrium through purging, effectively extracting a larger proportion of target analytes. This makes DHS particularly valuable for method development when analyzing new chemical entities with unknown binding characteristics.
Detailed Experimental Protocols
Static Headspace Method for Amine Analysis
Instrument Configuration and Reagents:
- GC System: Agilent 6890 with G1888 headspace autosampler and 20-mL sample vials [35]
- Column: Restek RTX-5 AMINE (30 m × 530 µm × 5 µm film thickness) [35]
- Diluents: DMSO, DMF, HPLC-grade water, NaOH solutions (concentration varies by analyte) [35]
- Standard Solutions: Prepared fresh from analytical-grade amines (triethylamine, n-butylamine, allylamine) [35]
Sample Preparation Protocol:
- Prepare stock solutions by diluting 138 µL triethylamine into 100 mL appropriate diluent
- Create analysis samples by diluting 1 mL stock solution into 100 mL with corresponding diluent (final concentration: 0.0100 mg/mL for TEA)
- For n-butylamine: dilute 213 µL into 250 mL diluent, then 1 mL into 25 mL (final concentration: 0.0252 mg/mL)
- For allylamine: dilute 300 µL into 100 mL diluent, then 1 mL into 100 mL (final concentration: 0.0228 mg/mL)
- Transfer 1 mL of final dilution to 20-mL headspace vial and immediately seal [35]
GC and Headspace Parameters:
- GC Method for TEA/n-butylamine: Initial 50°C (3 min hold), ramp 20°C/min to 130°C (2 min hold), 20°C/min to 170°C (3 min hold), 30°C/min to 240°C (4 min hold)
- Injector: 200°C; Detector (FID): 300°C; Helium flow: 3.8 mL/min constant
- Headspace Conditions: Oven: 80°C; Loop: 90°C; Transfer line: 100°C; Vial equilibration: 5-15 minutes; Pressurization: 0.5 min; Loop fill: 0.1 min; Loop equilibration: 0.05 min; Injection: 0.5 min [35]
ITEX-DHS Method for Complex Matrices
System Configuration:
- PAL System with ITEX-DHS attachment compatible with standard GC/MS systems [71]
- Sorbent trap selection based on analyte volatility (Tenax TA, carbon-based, or mixed beds)
- Standard 20-mL headspace vials with secure septa
Sample Preparation and Loading:
- Weigh appropriate sample amount (varies by matrix) directly into headspace vial
- For solid samples, consider addition of internal standard solution
- For complex matrices like honey or blood, minimal preparation required beyond weighing
- Seal vials immediately to prevent volatile loss
ITEX-DHS Operational Sequence:
- Condition sorbent trap according to manufacturer specifications
- Set incubation temperature and time based on sample matrix (typically 5-15 minutes)
- Configure extraction parameters: Number of extraction strokes (10-50 cycles), stroke speed, and volume
- Set desorption temperature and time based on analyte volatility and sorbent characteristics
- Implement GC/MS method appropriate for target analytes
Method Optimization Considerations:
- Number of extraction cycles directly correlates with sensitivity but increases method time
- Incubation temperature should be optimized to maximize vapor pressure without degrading samples
- Vacuum-assisted extraction (V-ITEX) may be employed for semi-volatile compounds [71]
- System performance should be validated with standards in relevant matrix
Workflow Visualization
Diagram 1: Method Selection Workflow for Headspace Techniques. The decision pathway begins with analyte characterization, directing high-concentration routine analysis toward Static Headspace and trace analysis in complex matrices toward Dynamic Headspace (ITEX-DHS).
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Headspace Method Development
| Reagent/Material | Function & Application | Performance Considerations |
|---|---|---|
| Restek RTX-5 AMINE Column | Specialized chromatography column for amine analysis | Minimizes peak tailing for basic compounds; 30 m × 530 µm × 5 µm configuration optimal for headspace [35] |
| DMSO (Dimethylsulfoxide) | High-boiling-point diluent for SHS | Reduces volatility of interfering compounds; improves amine response at 1 mL sample volume [35] |
| ITEX Sorbent Traps | Analyte enrichment in DHS | Multiple chemistries available; selection depends on analyte volatility and polarity [71] |
| NaOH Solutions (0.001-0.1M) | Basic diluent modifier | Enhances volatility of basic compounds like amines; concentration must be optimized per analyte [35] |
| 20-mL Headspace Vials | Standardized sample containers | Consistent vial dimensions critical for equilibration reproducibility; proper sealing essential [35] |
The comparative analysis reveals that Static Headspace and Dynamic Headspace serve complementary roles in modern analytical laboratories. SHS provides a robust, efficient solution for routine analysis of moderately volatile compounds where sensitivity requirements are within the parts-per-million range and sample throughput is prioritized. Its minimal sample preparation and established methodology make it ideal for quality control environments analyzing known compounds in consistent matrices [35].
DHS, particularly ITEX-based systems, offers superior capabilities for method development and trace analysis where sensitivity demands approach parts-per-billion levels or when analyzing complex, variable matrices. The enrichment capability, combined with vacuum-assisted options for semi-volatile compounds, provides analytical chemists with a powerful tool for challenging applications across pharmaceutical, environmental, and food science domains [71].
Strategic method selection should begin with careful assessment of sensitivity requirements, matrix complexity, and available instrumentation. For novel compounds, preliminary DHS analysis can establish detection limits and extraction efficiency, with potential migration to SHS methods once compounds are well-characterized and sensitivity requirements are confirmed. The ongoing automation of both techniques continues to reduce operational barriers, making sophisticated headspace analysis increasingly accessible to researchers across disciplines.
In pharmaceutical research and drug development, the analysis of residual solvents is a critical quality control step, governed by strict regulatory guidelines such as USP General Chapter <467> [35]. Headspace gas chromatography (HS-GC) has emerged as a premier technique for this application, allowing researchers to analyze volatile organic compounds without introducing complex sample matrices into the chromatographic system [48]. The fundamental principle behind headspace sampling involves analyzing the vapor phase in equilibrium with a solid or liquid sample in a sealed container, thus preventing non-volatile residues from accumulating in the GC inlet and column [48]. Within this field, two primary techniques dominate: static headspace and dynamic headspace (often referred to as purge and trap). This guide provides an objective comparison of these techniques, with particular focus on managing sample volume consistency and avoiding contamination—two fundamental factors directly impacting analytical reproducibility and data integrity in solvent research.
Technical Comparison: Static vs. Dynamic Headspace
Table 1: Fundamental Characteristics of Static and Dynamic Headspace Techniques
| Feature | Static Headspace GC | Dynamic Headspace GC |
|---|---|---|
| Basic Principle | Equilibrium-based sampling in a closed system [12] | Continuous purging with inert gas [12] |
| Sample Preparation | Minimal preparation required [12] | Requires setup for gas flow and trapping [12] |
| Sensitivity | Good for many volatiles [12] | Higher sensitivity for trace-level analysis [12] |
| Analysis Time | Longer equilibration time [12] | Generally faster analysis [12] |
| Primary Applications | Residual solvents, flavors, VOCs [12] | Trace analysis in water, air, solids [12] |
| System Complexity | Simpler setup [12] | More complex setup with traps and gas flow systems [12] |
| Risk of Contamination | Lower risk due to closed system [48] [12] | Potential for loss of volatiles or introduction of contaminants [12] |
Static headspace sampling operates on an equilibrium principle, where the sample is placed in a sealed vial and heated to promote the release of volatile compounds into the headspace. After a predetermined equilibration time, a portion of this gas is injected into the GC system [12]. This technique is particularly valued for its simplicity and effectiveness in preventing system contamination, as non-volatile materials remain in the vial [48]. In contrast, dynamic headspace employs a continuous flow of inert gas to purge volatiles from the sample, which are then trapped and concentrated on an adsorbent material before being thermally desorbed into the GC [48] [4]. This continuous extraction process provides enhanced sensitivity for trace-level compounds but involves more complex instrumentation [12].
Experimental Protocols for Reproducible Results
Optimizing Static Headspace Methods
A study analyzing residual solvents in β-cyclodextrin provides a robust protocol for static headspace optimization. The researchers systematically evaluated critical parameters to achieve high accuracy and precision [53]:
- Sample Injection Volume: The study compared 100 μL and 500 μL injection volumes, finding that 100 μL provided suitable accuracy while minimizing solvent use [53].
- Equilibration Conditions: Optimal conditions were determined to be 60°C for 45 minutes, balancing efficiency with complete release of volatiles [53].
- Matrix Modifiers: The investigation tested the effect of adding salts such as CaCl₂, finding it adversely affected recovery efficiency in this application, highlighting the need for application-specific optimization [53].
This optimized method yielded excellent results, with coefficients of determination (R²) exceeding 0.99 for target analytes (trichloroethylene and toluene), recovery rates between 91.7-106.0%, and relative standard deviation (RSD) of 1.0-8.9%, demonstrating high precision [53].
Sample Volume Consistency in Practice
Research on amine analysis reveals the complex relationship between sample volume and analytical signal. Contrary to conventional wisdom that larger sample volumes always increase response, studies demonstrated that increasing sample volume beyond an optimal point does not yield expected signal improvements [35]. When analyzing triethylamine, n-butylamine, and allylamine, sample volumes higher than 1 mL failed to produce the anticipated increase in signal strength [35]. This phenomenon occurs because increasing liquid sample volume directly reduces the headspace volume (VG) in the vial, altering the phase ratio (β = VG/V_S) and consequently affecting the equilibrium concentration of analytes in the headspace [48]. This underscores the critical importance of maintaining consistent sample volumes for reproducible results.
Contamination Control Strategies
Both static and dynamic headspace techniques offer contamination control benefits by preventing non-volatile matrix components from entering the GC system [48] [26]. However, each approach presents unique contamination risks:
- Static Headspace Contamination Control: The closed-system nature of static headspace minimizes opportunities for external contamination [12]. Key contamination control points include selecting septa that can withstand incubation temperatures without degrading and ensuring caps are properly crimped to prevent leaks [26].
- Dynamic Headspace Contamination Risks: The additional components in dynamic systems (traps, transfer lines, valves) introduce potential contamination points. The continuous gas flow may also lead to loss of highly volatile compounds if not properly controlled [12].
Essential Research Reagent Solutions
Table 2: Key Research Reagents and Materials for Headspace Analysis
| Item | Function | Application Notes |
|---|---|---|
| Headspace Vials | Provide sealed environment for sample equilibration [35] | 20-mL vials commonly used; ensure 50% headspace volume minimum [26] |
| PTFE/Silicone Septa | Maintain vial seal while allowing sample extraction [72] | Must withstand incubation temperatures without degradation [26] |
| Salting-Out Agents | Modify matrix to enhance volatile release [26] | Sodium chloride most common; NaOH or Na₂SO₄ may be better for specific matrices [26] [72] |
| High-Boiling Solvents | Dissolve samples without interfering with volatiles [35] | DMSO, DMF effectively dissolve various pharmaceutical compounds [35] |
| Internal Standards | Correct for analytical variability [72] | Deuterated analogs (e.g., 1,4-dioxane-d₈) ideal for compensation [72] |
| Polydimethylsiloxane (PDMS) | Sorptive polymer for dynamic sampling [73] | Apolar polymer that absorbs lipophilic compounds; used in tubing or SPME fibers [73] |
Workflow and Pathway Visualization
The following workflow diagrams illustrate the key processes in static and dynamic headspace analysis, highlighting critical control points for volume consistency and contamination prevention.
Headspace Analysis Critical Control Pathways
The pathways above visualize the operational steps for both techniques, highlighting critical control points where volume consistency and contamination prevention must be managed. In static headspace, the sample preparation and equilibration stages are particularly crucial for maintaining reproducible phase ratios [48] [26]. For dynamic headspace, the gas purging and trapping phases present potential contamination risks that require careful control [12].
The selection between static and dynamic headspace techniques represents a critical methodological decision in solvent research, with significant implications for reproducibility, data quality, and regulatory compliance. Static headspace offers simplicity, reduced contamination risk, and sufficient sensitivity for many pharmaceutical applications, particularly residual solvent analysis [12] [35]. Dynamic headspace provides enhanced sensitivity for trace-level analysis but requires more complex instrumentation and presents additional contamination control challenges [12]. For both techniques, rigorous attention to sample volume consistency, temperature control, and materials integrity forms the foundation of reproducible results. By implementing the optimized protocols and control strategies outlined in this guide, researchers and drug development professionals can ensure data integrity while advancing their solvent research objectives.
Head-to-Head Comparison: Validating Sensitivity, Speed, and Regulatory Fit
In the analysis of volatile compounds, particularly for residual solvents in pharmaceuticals or contaminants in environmental samples, static and dynamic headspace sampling are two cornerstone techniques. Static headspace (S-HS) is celebrated for its simplicity and robustness, while dynamic headspace (D-HS), often referred to as purge and trap, is renowned for its superior sensitivity. Selecting the appropriate method is a critical decision that directly impacts data quality, operational efficiency, and cost. This guide provides a direct, data-driven comparison of these two techniques to inform researchers, scientists, and drug development professionals in their analytical method selection.
Core Principles at a Glance
The fundamental difference between these techniques lies in how the volatile analytes are transferred from the sample to the analytical instrument.
- Static Headspace (S-HS): The sample is placed in a sealed vial and heated to achieve equilibrium between the sample matrix and the headspace gas above it. A single aliquot of this gas is then injected into the Gas Chromatograph (GC) [35] [21].
- Dynamic Headspace (D-HS): An inert gas continuously purges the sample, stripping volatile compounds. These analytes are focused onto a sorbent trap. After purging, the trap is heated to rapidly desorb the concentrated analytes into the GC [4] [71].
Direct Technique Comparison
The table below summarizes the key performance characteristics and operational factors of static and dynamic headspace methods.
| Feature | Static Headspace (S-HS) | Dynamic Headspace (D-HS / Purge & Trap) |
|---|---|---|
| Sensitivity & Detection Limits | Moderate sensitivity. Detection limits typically in the parts-per-billion (ppb) range for many compounds [33]. | Very high sensitivity. Provides 50–100x greater signal for many analytes compared to S-HS, achieving detection in the parts-per-trillion (ppt) range [15] [33]. |
| Complexity & Workflow | Simple, straightforward workflow with minimal steps. Easy to automate and highly robust for routine use [15] [23]. | More complex setup involving a purge system, sorbent trap, and desorption components. Requires optimization of more parameters [4]. |
| Analysis Speed & Throughput | Fast cycle times. Typical equilibration times are 5-15 minutes, enabling high sample throughput [35]. | Slower per sample due to longer purge and desorb cycles. Throughput is lower but can be offset by high sensitivity reducing re-analysis needs [4]. |
| Cost & Instrumentation | Lower initial instrument cost and lower operational complexity. Consumes less gas and requires less maintenance [34]. | Higher initial investment. Ongoing costs include sorbent traps and potentially more carrier gas [34] [62]. |
| Ideal Application Scope | Ideal for routine analysis of volatile and semi-volatile compounds in simple to moderately complex matrices where high sensitivity is not critical (e.g., USP <467> residual solvents) [74] [23]. | Essential for trace-level analysis of volatile organic compounds (VOCs) in complex matrices (e.g., environmental water analysis, flavor profiling, impurity identification) [33] [4]. |
| Matrix Effects | Can be significant, as the partition coefficient (K) between the sample and gas phase is critical [21]. | Continuous removal of analytes can reduce matrix effects, leading to more efficient extraction from difficult samples [15] [4]. |
Experimental Protocols in Practice
Protocol 1: Static Headspace for Pharmaceutical Residual Solvents
This protocol is adapted from a platform method for determining 27 residual solvents in active pharmaceutical ingredients (APIs) [74] [23].
- Sample Preparation: Accurately weigh ~100 mg of API into a headspace vial. Add 1 mL of a high-boiling diluent like N-Methyl-2-pyrrolidone (NMP) or dimethyl sulfoxide (DMSO). Seal the vial immediately with a PTFE-lined crimp cap [74] [23].
- Instrumental Conditions:
- HS Sampler: Oven temperature: 80-120°C; Needle temperature: 110-130°C; Transfer line temperature: 115-135°C; Equilibration time: 5-15 minutes; Withdrawal time: 0.2-1.0 minute [35] [23].
- GC: Inlet: Split (40:1), 200°C; Column: Mid-polarity fused silica capillary (e.g., DB-624, 30 m x 0.32 mm, 1.8 µm); Oven Program: Ramp from 40°C to 240°C; Detector: Flame Ionization Detector (FID) at 300°C [74] [23].
- Key Experimental Data: This method demonstrated excellent precision (Relative Standard Deviation typically <15.0%) and achieved Limits of Quantitation (LOQs) meeting ICH Q3C guidelines for Class 2 and 3 solvents. Sample and standard solutions were stable for at least 10 days at room temperature [74].
Protocol 2: Dynamic Headspace for Trace VOC Analysis in Water
This protocol is based on EPA Method 8260, which uses dynamic headspace for analyzing volatile organic compounds in various matrices [33].
- Sample Preparation: Transfer 5 mL of aqueous sample into a 22 mL headspace vial and seal. For solid samples, a smaller weight is used with an appropriate amount of water [33].
- Instrumental Conditions:
- D-HS (Purge & Trap) Sampler: Purge time: 11 minutes with helium; Purge gas flow: 40 mL/min; Desorb preheat: 240°C; Desorb time: 1 minute at 260°C; Trap: A sorbent trap such as a "Number 9 trap" which is a common multi-bed sorbent [33].
- GC-MS: Column: FactorFour VF-624ms (20 m x 0.15 mm, 0.84 µm); Oven Program: Ramp from 35°C to 240°C; Detector: Mass Spectrometer (MS) in scan mode (e.g., 35-280 m/z) [33].
- Key Experimental Data: In a direct comparison, the dynamic mode provided detection limits of 0.5 ppb, while the static mode achieved 10 ppb for the same standard. The dynamic method produced peak areas 20 to 125 times greater for various VOCs compared to the static mode [33].
The Scientist's Toolkit: Essential Research Reagents and Materials
| Item | Function & Application |
|---|---|
| High-Boiling Solvents (NMP, DMSO, DMF) | Used as diluents in S-HS to dissolve sample matrices and adjust the partition coefficient (K), enhancing the release of volatile analytes into the headspace [35] [23]. |
| Sorbent Trap | A critical component in D-HS. Contains materials (e.g., Tenax, charcoal, silica) that adsorb VOCs during the purge cycle and release them upon thermal desorption. Trap selection is analyte-dependent [33] [71]. |
| Headspace Vials | Sealed vials (typically 10-22 mL) designed to withstand pressure and maintain a gas-tight seal during heating and sampling [35]. |
| Custom Stock Standard | A premade mixture of target solvents at known concentrations. This ensures analytical accuracy, improves lab efficiency, and is essential for method validation and quantitation [74] [23]. |
| Ionic Liquids | Emerging as advanced, environmentally friendly ("green") diluents for headspace GC. They can improve the analysis of very low vapor pressure compounds and offer unique selectivity [74] [4]. |
The choice between static and dynamic headspace is a trade-off between simplicity and supreme sensitivity.
- For high-throughput, routine analyses where target analytes are relatively abundant and matrices are well-understood, static headspace offers a robust, cost-effective, and simple solution. Its widespread adoption in pharmaceutical quality control (e.g., USP <467>) is a testament to its reliability [74] [23].
- For challenging applications requiring trace-level detection, such as identifying unknown impurities, monitoring environmental pollutants, or characterizing food aromas, dynamic headspace is the unequivocal choice. Its powerful pre-concentration capability provides the necessary sensitivity, despite greater method complexity and cost [15] [33] [4].
Understanding these core differences enables scientists to make an informed decision, aligning their analytical strategy with specific research and development goals.
In the quality control of pharmaceuticals, the analysis of residual solvents is a critical safety requirement, governed by strict regulatory guidelines such as ICH Q3C and USP 〈467〉. These volatile organic compounds, used in the manufacturing of Active Pharmaceutical Ingredients (APIs), must be monitored to ppm or ppb levels due to their inherent toxicity [75] [76]. Headspace Gas Chromatography (HS-GC) has emerged as the gold standard technique for this analysis, primarily because it introduces a clean vapor sample into the GC system, avoiding contamination from non-volatile matrix components [12] [77].
The two principal sampling approaches for this analysis are static headspace (S-HS) and dynamic headspace (D-HS), also known as purge and trap. The choice between them is often dictated by the required sensitivity and the complexity of the sample matrix. This guide provides an objective, data-driven comparison of their performance, focusing on Limits of Detection (LOD) and operational characteristics, to aid researchers and scientists in selecting the most appropriate method for their trace-level residual solvent testing.
At its core, headspace sampling analyzes the volatile compounds present in the gas phase (the headspace) above a solid or liquid sample in a sealed vial [4]. The fundamental differences between the static and dynamic modes lie in how this vapor phase is sampled and introduced into the gas chromatograph.
- Static Headspace (S-HS) is an equilibrium-based technique. The sample is heated in a sealed vial until the volatile compounds partition between the sample matrix and the headspace gas, reaching a state of equilibrium. Once achieved, a portion of this headspace gas is extracted and injected into the GC system [12] [4]. Its simplicity and minimal sample preparation make it a widely adopted approach.
- Dynamic Headspace (D-HS), or purge and trap, is a non-equilibrium, exhaustive extraction technique. An inert gas, such as nitrogen or helium, continuously purges the sample, sweeping volatile compounds from the headspace onto a sorbent trap. The trapped analytes are then thermally desorbed and transferred to the GC, leading to a significant pre-concentration effect [12] [4] [36].
The table below summarizes the key characteristics of both techniques, providing a high-level comparison of their operational principles.
Table 1: Core Characteristics of Static and Dynamic Headspace Techniques
| Feature | Static Headspace (S-HS) | Dynamic Headspace (D-HS) |
|---|---|---|
| Basic Principle | Equilibrium-based sampling | Continuous purging with inert gas (exhaustive) |
| Sensitivity | Good for many volatiles | Higher sensitivity; suitable for trace-level analysis |
| Analysis Time | Longer equilibration time required | Generally faster analysis |
| Sample Preparation | Minimal preparation required | Requires setup for gas flow and trapping |
| Instrument Complexity | Simpler setup | More complex setup with trapping system |
| Best For | Relatively simple matrices; volatile targets | Complex matrices; trace-level and low-volatility analytes |
Quantitative Performance Comparison: LOD and Sensitivity
The most significant performance differentiator between S-HS and D-HS is their achievable sensitivity, directly impacting the Limit of Detection (LOD). The pre-concentration step inherent to dynamic headspace provides a substantial advantage for detecting analytes at ultra-trace levels.
A systematic comparison of six automated headspace techniques, categorized into static sampling, static enrichment (like SPME), and dynamic enrichment, quantified this performance gap. The study evaluated Method Detection Limits (MDLs) and extraction yields for a common set of Volatile Organic Compounds (VOCs) [16].
Table 2: Quantitative Performance Metrics from Systematic Comparison
| Technique Category | Example Techniques | Typical Extraction Yield | Achievable Method Detection Limit (MDL) |
|---|---|---|---|
| Static Sampling | Syringe, Loop | ~10-20% | Approx. 100 ng/L (ppt) |
| Static Enrichment | SPME, PAL SPME Arrow | Up to ~80% | Picogram per liter (ppq) range |
| Dynamic Enrichment | ITEX, Trap Sampling (D-HS) | Up to ~80% | Picogram per liter (ppq) range |
The data shows that while static sampling techniques offer sufficient sensitivity for many applications (around 100 ng/L), both static and dynamic enrichment techniques can achieve extraction yields up to 80%, pushing detection limits into the picogram-per-liter range [16]. This makes D-HS indispensable for detecting Class 1 solvents, such as benzene, where regulatory limits are exceptionally low, often requiring LODs as low as 0.5 ppm [75].
Experimental Protocols and Methodologies
A Representative Static Headspace (S-HS) Protocol
A 2025 study on the analysis of 12 residual solvents in Avibactam sodium API provides a robust, validated example of a S-HS method. The method was optimized for separation and sensitivity, complying with pharmacopeial standards [77].
- Instrumentation: Agilent GC system (7890B) with a Flame Ionization Detector (FID) and an auto-headspace sampler (7694A). A DB-624UI capillary column (30 m × 0.32 mm × 1.8 µm) was used.
- Sample Preparation: An internal standard solution (Isopropyl acetate, IPAC, 0.1 mg/mL) in N-methylpyrrolidone (NMP) was used. The API sample (0.2 g) was dissolved in 2 mL of this internal standard solution.
- Headspace Conditions:
- Equilibrium Temperature: 80 °C
- Equilibrium Time: 30 minutes
- Loop Temperature: 90 °C
- GC-FID Conditions:
- Carrier Gas: Nitrogen at 2.0 mL/min
- Oven Program: 40 °C (hold 5 min) to 120 °C at 20 °C/min (hold 2 min), then to 200 °C at 20 °C/min (hold 5 min).
- Detector Temperature: 250 °C
- Validation Results: The method demonstrated excellent linearity (R² ≥ 0.99) for all 12 solvents, with precision within acceptable limits and average recovery rates confirming accuracy [77].
A Representative Dynamic Headspace (D-HS) Optimization Protocol
A 2022 study established a generalized procedure for optimizing D-HS extractions using Design of Experiments (DoE), a multivariate approach superior to the traditional one-factor-at-a-time method. This is crucial because D-HS has multiple interacting parameters [36].
- Instrumentation: Automated Dynamic Headspace Module with a Thermal Desorption Unit (TDU) and a programmable-temperature vaporization (PTV) inlet coupled to a GC×GC–TOF-MS. A Tenax TA sorbent trap was used.
- DoE Optimization:
- Factors: The three critical factors optimized were Incubation Time, Purge Flow Rate, and Purge Flow Volume.
- Design: A Box-Behnken design with three factors was used to model the response surface with a minimal number of experimental runs.
- Response: The methods were optimized based on the total number of detected compounds and the total summed peak area.
- DHS and Desorption Parameters:
- Purge Gas: High-purity nitrogen.
- Trap Temperature: 25 °C for trapping volatiles.
- Desorption: Trapped analytes were thermally desorbed from the trap at 250 °C and cryofocused in the PTV at -100 °C before GC analysis.
The following workflow diagram illustrates the logical sequence and key parameter choices for developing a D-HS method, from sample preparation to data analysis.
Diagram: D-HS Method Development Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Successful and compliant residual solvent analysis relies on specific materials and reagents. The following table details essential items used in the featured experiments and their critical functions [77] [76] [78].
Table 3: Essential Materials and Reagents for Residual Solvent Analysis
| Item | Function & Rationale |
|---|---|
| DB-624 / DB-624UI Capillary Column | A mid-polarity, bonded 6% cyanopropylphenyl / 94% dimethyl polysiloxane column. Standard for volatile compound separation as per USP 〈467〉; ideal for residual solvents. |
| N-Methylpyrrolidone (NMP) | A high-boiling-point, water-miscible solvent. Used to dissolve sample matrices (APIs) without interfering in the volatile analysis; helps create a consistent sample solution. |
| Internal Standards (e.g., IPAC) | A compound not present in the sample, added at a known concentration. Corrects for minor variations in sample preparation and injection; crucial for achieving quantitative accuracy. |
| Dimethyl Sulfoxide (DMSO) / Dimethylformamide (DMF) | High-boiling-point dilution solvents. Alternative to water for dissolving APIs with poor aqueous solubility; headspace sensitivity can vary significantly with the choice of dilution solvent. |
| Tenax TA Sorbent Trap | A sorbent material (polymer of 2,6-diphenylene oxide) with high thermal stability. Used in D-HS to trap and concentrate volatiles purged from the sample before thermal desorption into the GC. |
The choice between static and dynamic headspace for residual solvent analysis is not a matter of one technique being universally superior, but rather of selecting the right tool for the specific analytical requirement.
- Static Headspace (S-HS) is the method of choice for routine quality control where target solvents are relatively volatile and present at concentrations above approximately 100 ng/L. Its simplicity, robustness, and compliance with pharmacopeial methods make it ideal for high-throughput environments analyzing known solvents in standard APIs [12] [77].
- Dynamic Headspace (D-HS) is unequivocally more powerful for trace-level analysis and method development for complex or unknown volatile profiles. Its pre-concentration capability provides a significant sensitivity advantage, achieving LODs in the picogram-per-liter range, which is essential for monitoring toxic Class 1 solvents or analyzing challenging matrices [36] [16].
For drug development professionals, this comparison underscores that S-HS offers efficiency for established, well-defined applications, while D-HS provides the necessary firepower for pushing the boundaries of sensitivity and tackling the most complex analytical challenges in modern pharmaceutical development.
Static Headspace (SHS) and Dynamic Headspace (DHS) are two established techniques for extracting volatile compounds from complex samples prior to gas chromatography. While SHS is renowned for its operational simplicity and ease of automation, DHS provides superior sensitivity and lower detection limits, making it suitable for trace-level analysis. The choice between them involves a direct trade-off between analysis throughput and analytical sensitivity, which this guide explores through experimental data and workflow considerations.
Technical Comparison: Static vs. Dynamic Headspace
The following table summarizes the core characteristics of SHS and DHS that impact laboratory throughput and efficiency.
Table 1: Key Characteristics Comparison of Static and Dynamic Headspace Techniques
| Feature | Static Headspace (SHS) | Dynamic Headspace (DHS) |
|---|---|---|
| Fundamental Principle | A single aliquot of the equilibrated headspace is extracted and injected. [3] [53] | Continuous gas flow purges volatiles from the sample, which are trapped and concentrated on an adsorbent material. [3] [71] |
| Primary Throughput Driver | Shorter cycle times; simpler, highly automatable workflow. [53] | Longer extraction times but higher sensitivity can reduce or eliminate need for re-analysis. [79] |
| Typical Analysis Time | Faster for individual samples (e.g., ~45 min equilibration). [53] | Slower per sample due to purge, trap, and desorb cycles. [71] |
| Automation Potential | Excellent; readily integrated with commercial autosamplers (e.g., PAL, GERSTEL MPS). [3] [80] | Excellent for modern systems (e.g., ITEX, DHS); can be fully automated. [71] |
| Sensitivity / Detection Limits | Good, but limited by single-step extraction. [81] | Excellent; high concentration capacity yields dramatically lower detection limits. [81] [79] |
| Sample Throughput | High for a given number of samples; ideal for quantitative, high-volume routine analysis. [80] | Can be lower per sample, but superior sensitivity may enable fewer dilutions/re-runs for complex matrices. |
| Method Development | Straightforward; fewer parameters to optimize (e.g., temp, time, vial size). [7] | More complex; requires optimization of purge flow, trap type, and desorption conditions. [3] |
Experimental Data and Performance Metrics
Throughput and Sensitivity in Pharmaceutical Analysis
A study on residual solvents in the drug substance Suvorexant demonstrated the application of automated SHS. The method provided satisfactory resolution for eight residual solvents, including n-heptane, with linearity (r > 0.990) and relative standard deviations (RSD) below 5.0%, demonstrating the precision achievable with automated SHS for quality control. [76] This showcases SHS's capability for high-throughput, reliable analysis in regulated environments.
Quantitative Throughput Advantage in Direct MS Analysis
While not a headspace technique, Selected Ion Flow Tube Mass Spectrometry (SIFT-MS) provides a compelling case for how speed transforms workflow efficiency. One study highlighted that its chromatography-free runtime of under 5 minutes allowed for a 2.9-fold increase in throughput and a 1.8-fold faster time to the first result compared to GC-MS methods when using the method of standard additions. [80] This principle underscores the throughput benefit of simpler, faster vapor introduction systems.
Optimization for Speed and Reproducibility in Environmental Monitoring
A robust, statistically validated SHS method for volatile petroleum hydrocarbons (VPHs) in water used an experimental design to optimize parameters. [7] The optimized method emphasized full automation, reduced solvent consumption, and enhanced reproducibility, key factors for efficient, high-throughput environmental monitoring workflows. [7]
Detailed Experimental Protocols
Protocol 1: Static Headspace-GC-MS for Residual Solvents in a Solid Matrix
This protocol is adapted from a study detecting residual trichloroethylene and toluene in β-cyclodextrin. [53]
- 1. Sample Preparation: An exact amount of solid sample (e.g., β-cyclodextrin) is weighed directly into a headspace vial. The vial is immediately sealed with a PTFE/silicone septum and an aluminum crimp cap to prevent analyte loss.
- 2. Equilibration: The sealed vials are placed in the headspace autosampler oven and heated at 60°C for 45 minutes to allow the volatile solvents to partition into the headspace. The study found that the addition of salt (CaCl₂) adversely affected recovery and is omitted. [53]
- 3. Automated Injection: After equilibration, the autosampler extracts a defined volume of headspace vapor (e.g., 100 μL) from the vial using a gas-tight syringe and injects it into the GC inlet.
- 4. GC-MS Analysis: Separation is typically performed on a mid-polarity capillary column (e.g., DB-624). The oven temperature is ramped to separate the target solvents, which are detected and quantified by mass spectrometry.
Protocol 2: In-Tube Extraction Dynamic Headspace (ITEX-DHS)
This protocol describes a modern, automated approach to DHS. [71]
- 1. System Setup: A DHS device, such as the ITEX, is installed on a robotic autosampler. The system uses a gas-tight syringe with a needle containing a packed adsorbent trap.
- 2. Headspace Extraction (Purge & Trap): The sample vial is heated and agitated. The syringe repeatedly aspirates and dispenses the headspace vapor (typically 10-50 extraction cycles), forcing the volatile compounds through the needle where they are trapped on the adsorbent.
- 3. Desorption and Injection: After the extraction cycles, the trapped analytes are thermally desorbed from the adsorbent inside the needle by heating the syringe. The concentrated analytes are then injected directly into the GC inlet in a single, narrow band.
- 4. GC Analysis: The concentrated analytes are separated and detected, yielding higher sensitivity than a single SHS injection.
Workflow and Automation Diagrams
The following diagram illustrates the automated workflows for both SHS and DHS techniques, highlighting the steps that impact throughput.
Figure 1: Automated Workflow Comparison. SHS offers a simpler, faster path, while DHS includes extra steps for higher sensitivity. [3] [71] [53]
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Materials and Equipment for Headspace Analysis
| Item | Function / Application | Example from Literature |
|---|---|---|
| PAL or GERSTEL MPS Autosampler | Automated, multi-purpose robotic systems for unattended SHS, DHS, SPME, and liquid injection, enabling high-throughput workflow integration. [3] [80] | Used for high-throughput analysis of volatile impurities in consumer products via the method of standard additions. [80] |
| ITEX (In-Tube Extraction) Device | An automated system for performing Dynamic Headspace sampling; it concentrates volatiles via repeated headspace extraction through an adsorbent-packed needle. [71] | Applied in the analysis of flavors, pesticides, and biomarkers directly from complex matrices like honey, cheese, and blood. [71] |
| Headspace Vials & Seals | Sealed containers (typically 10-20 mL) with PTFE/silicone septa and aluminum caps that maintain a closed system to prevent volatile loss during incubation. [7] [53] | Critical for all protocols to ensure no analyte escapes during the heating/equilibration step, guaranteeing quantitative accuracy. [7] [53] |
| DB-624 Capillary GC Column | A mid-polarity, bonded-phase column specifically designed for the separation of volatile organic compounds, including residual solvents. [76] | Used for the separation and analysis of eight residual solvents in the pharmaceutical product Suvorexant. [76] |
| Sorbent Traps (for DHS) | Adsorbent materials (e.g., Tenax, carbon-based sorbents) packed inside a needle or tube to trap and concentrate volatiles during the purge phase of DHS. [3] [71] | The core component enabling the high concentration capability of DHS and ITEX techniques. [3] [71] |
| Enhanced Matrix Removal (EMR) Cartridges | Solid-phase extraction cartridges designed for selective removal of matrix interferences like lipids and pigments from complex samples prior to analysis. [82] | Used for pass-through cleanup in PFAS analysis in food, simplifying workflow and reducing manual sample prep time. [82] |
The accurate quantification of residual solvents is a critical requirement in pharmaceutical development, mandated by regulatory standards such as the International Council for Harmonisation (ICH) Q3C guidelines and the United States Pharmacopeia (USP) General Chapter <467> [83]. These regulations aim to limit patient exposure to potentially harmful solvents used in manufacturing processes. For researchers and drug development professionals, selecting the appropriate analytical technique is paramount for generating valid, submission-ready data. Headspace gas chromatography (HS-GC) has emerged as the principal technique for this analysis, primarily implemented in two forms: static headspace (SHS) and dynamic headspace (DHS), also known as purge and trap.
This guide provides a objective comparison of static and dynamic headspace techniques within the framework of regulatory method validation. It examines the fundamental principles, relative performance characteristics, and specific applicability to meeting the requirements of ICH guidelines and USP <467>, providing scientists with the data necessary to make an informed choice for their analytical methods.
Fundamentals of Headspace Extraction Techniques
Static Headspace (SHS) Extraction
Static Headspace Extraction is an equilibrium-based technique where a sample is placed in a sealed vial and heated until the volatile compounds distribute between the sample matrix and the gas phase (headspace) [10] [4]. Once equilibrium is established, an aliquot of the vapor is collected from the headspace, typically using a gas-tight syringe or an automated valve system, and injected into the GC for analysis [10]. The concentration of an analyte in the headspace is governed by its partition coefficient (K), defined as the ratio of its concentration in the sample phase to its concentration in the gas phase at equilibrium [10].
Key Method Development Parameters for SHS:
- Temperature: Increased temperature favors the transfer of analytes to the vapor phase, thereby increasing signal response. However, the temperature must be controlled to avoid solvent vaporization or analyte degradation [10].
- Phase Ratio (β): The ratio of the vapor phase volume to the sample volume in the vial. A smaller phase ratio (larger sample volume) can enhance sensitivity for volatile analytes [10].
- Equilibrium Time: Ensuring the system reaches a state of dynamic equilibrium is critical for reproducibility [10].
Dynamic Headspace (DHS) Extraction
Dynamic Headspace Extraction, or purge and trap, is a non-equilibrium, exhaustive technique [10] [4]. An inert gas, such as helium or nitrogen, is continuously passed through (or over) the sample, stripping volatile compounds from the matrix. The purged analytes are then trapped and concentrated on a sorbent trap. After the purging process, the trap is heated and the analytes are desorbed into the GC for analysis [10] [4]. This continuous removal of volatiles, driven by LeChatelier's Principle, allows for a more complete extraction of analytes from the sample [10].
Key Method Development Parameters for DHS:
- Purge Gas Flow Rate and Time: These determine the volume of gas passed through the sample and impact the efficiency of analyte removal.
- Trap Sorbent: The selection of sorbent material is critical for efficiently trapping a wide range of volatiles while allowing for efficient thermal desorption.
- Desorption Temperature and Time: These must be optimized to ensure complete transfer of analytes from the trap to the GC without causing degradation.
The following workflow diagram illustrates the fundamental procedural differences between these two techniques.
Technical and Performance Comparison
The choice between SHS and DHS has significant implications for the sensitivity, scope, and efficiency of an analytical method. The following table summarizes the core performance characteristics of each technique, which are critical for method selection in a regulated environment.
Table 1: Performance Comparison of Static vs. Dynamic Headspace
| Parameter | Static Headspace (SHS) | Dynamic Headspace (DHS/Purge & Trap) |
|---|---|---|
| Fundamental Principle | Equilibrium partitioning [10] | Exhaustive extraction (non-equilibrium) [10] |
| Typical Sensitivity | High part-per-billion (ppb) to ppm [10] | Part-per-trillion (ppt) to ppb [10] [4] |
| Analyte Recovery | Partial (governed by K) [10] | Near-complete (qualitative removal) [10] |
| Best Suited For | Routine analysis of relatively volatile compounds at higher concentrations [10] | Ultratrace analysis, low volatility compounds, or complex matrices [10] [4] |
| Throughput | High (fast equilibrium and injection) | Moderate to Low (longer purge and desorb times) |
| Matrix Effects | Can be significant; requires careful optimization [10] | Reduced by exhaustive nature, but trapping efficiency is key |
| Regulatory Use Case | Standard residual solvent testing per USP <467> [83] | Analysis of very low-level genotoxic impurities or contaminants [4] |
| Ease of Method Development | Straightforward, well-understood parameters [10] | More complex, requires optimization of purge, trap, and desorb [4] |
Interpreting Experimental Data for Method Selection
When examining experimental data for regulatory submission, the following points are crucial:
- For SHS: The data should demonstrate that equilibrium has been robustly established and that the method is unaffected by minor variations in sample volume (phase ratio), particularly for volatile analytes [10]. The use of an internal standard is highly recommended to control for variations in matrix effects.
- For DHS: The focus should be on demonstrating quantitative recovery from the sample and from the trap. Data should validate that the chosen purge time and flow rate are sufficient for the analytes of interest and that no carryover or breakthrough occurs on the sorbent trap [4].
Meeting Regulatory Requirements: ICH and USP <467>
USP General Chapter <467> provides the enforceable standards for residual solvents testing in official USP articles. Its purpose is to "limit the amount of solvent that patients receive" [83]. Key points include:
- Applicability: It applies to all drug substances, excipients, and finished drug products covered by a USP or NF monograph, including existing commercial products [83].
- Testing Options: Manufacturers have the option to test the final drug product or all individual components (active ingredients and excipients) [83].
- Solvent Classification: It adopts the ICH Q3C classification of solvents into Class 1 (to be avoided), Class 2 (to be limited), and Class 3 (low toxic potential) [83].
Method Validation Considerations
Under the USP General Notices, the use of appropriately validated alternative methods is permitted, providing flexibility beyond the official methods described in the chapter [83]. Whether using a compendial or a validated in-house method, the validation must demonstrate:
- Specificity: The method must be able to unequivocally identify and quantify the target solvents in the presence of other components. For GC, this typically requires resolution from interfering peaks. USP notes that Procedures A and B are orthogonal and can be used to resolve co-elutions [83].
- Accuracy and Precision: For quantitative procedures (like USP <467> Procedure C), accuracy should be established using spiked samples, and precision (repeatability) must be demonstrated [83].
- Quantitation Limits: The method must be sufficiently sensitive to detect and quantify solvents at or below the specified limits. DHS may be necessary to achieve the required limits for certain Class 2 solvents in complex matrices.
- Robustness: The method should be resilient to small, deliberate variations in method parameters (e.g., equilibrium temperature, purge time, vial pressure).
The following diagram outlines the logical decision pathway for selecting an analytical approach that complies with USP <467>.
Alignment with ICH Q8(R2) and Quality by Design
The ICH Q8(R2) guideline emphasizes a systematic approach to pharmaceutical development, known as Quality by Design (QbD) [84] [85]. This approach can be applied to analytical method development:
- Defining an Analytical Target Profile (ATP): The ATP defines the required quality of the analytical data (e.g., sensitivity, precision) needed to control the product quality.
- Risk Assessment: Tools can be used to identify critical method parameters (e.g., equilibrium temperature in SHS, purge time in DHS) that impact the ATP.
- Design Space: A multidimensional combination and interaction of method parameters that have been demonstrated to provide assurance of quality. For SHS, this could involve establishing proven acceptable ranges for temperature, equilibration time, and sample volume [10].
- Control Strategy: This includes the validated procedure and system suitability tests to ensure the method remains in a state of control throughout its lifecycle.
Essential Research Reagent Solutions
The following table details key materials and reagents required for developing and validating headspace methods for residual solvents analysis.
Table 2: Essential Research Reagent Solutions for Headspace Analysis
| Item | Function/Description | Application Notes |
|---|---|---|
| Gas-Tight Syringe | Manual injection of headspace vapor from a sealed vial [10]. | For simple, non-automated SHS setup. Requires careful temperature control for reproducibility. |
| HS Autosampler | Automated system for vial heating, pressurization, and sample transfer to GC [10]. | Essential for high-throughput, reproducible analysis in a regulated lab. |
| Certified Reference Standards | Pure, certified materials for target solvents for calibration and quantification. | Required for method validation and ongoing system suitability testing. |
| Internal Standards | Stable, volatile compounds (e.g., dioxane, acetonitrile-d3) added to samples. | Corrects for volumetric inaccuracies and matrix effects, improving accuracy and precision. |
| Sorbent Traps | Cartridges containing materials (e.g Tenax, carbograph) for trapping volatiles in DHS [4]. | Trap selection is critical for analyte focus and must be validated for recovery and lack of carryover. |
| Inert Purge Gas | High-purity helium or nitrogen for DHS. | Used to strip volatiles from the sample matrix. |
| Appropriate Diluents | Solvents like dimethyl sulfoxide (DMSO), dimethylformamide (DMF), or water [4]. | Must dissolve the sample and not interfere with the analysis of target solvents. |
Both static and dynamic headspace techniques offer distinct advantages for residual solvents analysis in pharmaceutical products. Static Headspace is a robust, simple, and high-throughput equilibrium technique that is perfectly suited for routine compliance testing with USP <467> for most applications, especially when target solvents are relatively volatile and present at higher concentrations. In contrast, Dynamic Headspace is an exhaustive extraction technique that provides superior sensitivity and is the method of choice for ultratrace analysis, for compounds with low volatility, or in complex matrices where high preconcentration is needed.
The decision between SHS and DHS should be guided by a thorough understanding of the analytical target profile, which includes the required sensitivity, the physicochemical properties of the analytes, and the complexity of the sample matrix. By applying the principles of ICH Q8(R2) and Quality by Design to method development, and by rigorously validating the chosen method—whether compendial or alternative—against the requirements of USP <467>, scientists can ensure the generation of reliable, high-quality data that meets regulatory expectations for submission and patient safety.
The analysis of residual solvents in pharmaceutical products is a critical quality control step, ensuring patient safety by limiting potentially harmful organic volatile impurities. Headspace gas chromatography (HS-GC) is the established technique for this application, primarily implemented in two forms: static and dynamic headspace. Static headspace sampling involves heating a sample in a sealed vial until the volatile compounds reach equilibrium between the sample matrix and the gas phase (headspace) above it, after which a portion of this gas is injected into the GC system [12] [48]. In contrast, dynamic headspace (often called "purge and trap") continuously purges the sample with an inert gas, sweeping volatile compounds onto a trap where they are concentrated before being thermally desorbed into the GC [12] [86].
For regulatory testing of residual solvents in pharmaceuticals, static headspace is the prescribed method in the United States Pharmacopeia (USP) general chapter <467> [87]. This case study explores the development and validation of a static headspace GC-MS method for the simultaneous identification and quantitation of Class 1 and Class 2 solvents, highlighting its performance and practical utility for drug development professionals. The primary advantage of static headspace is its simplicity and minimal sample preparation, which reduces the risk of contamination from non-volatile components [12] [48]. However, it can have longer equilibration times and may offer limited sensitivity for certain trace-level analytes compared to dynamic methods [12] [14].
Method Comparison: Static vs. Dynamic Headspace
The choice between static and dynamic headspace sampling is dictated by the analytical requirements, sample matrix, and regulatory guidelines. The following table summarizes the key differences between these two techniques.
Table 1: Comparative Overview of Static and Dynamic Headspace GC
| Feature | Static Headspace GC | Dynamic Headspace GC |
|---|---|---|
| Basic Principle | Equilibrium-based sampling from the gas phase in a closed vial [12] [86] | Continuous purging of the sample with inert gas and trapping of volatiles [12] [86] |
| Sample Preparation | Minimal preparation required [12] | Requires setup for gas flow and trapping [12] |
| Sensitivity | Good for many volatiles [12] | Higher sensitivity, suitable for trace-level analysis [12] [86] |
| Analysis Time | Longer equilibration time to reach equilibrium [12] | Generally faster analysis [12] |
| Typical Applications | Residual solvents in pharmaceuticals, flavors, VOCs in food and environmental samples [12] [87] | Trace volatiles in water, air, and solid samples [12] [88] |
| System Complexity | Simpler setup [12] | More complex, requires traps and gas flow systems [12] [14] |
| Regulatory Examples | USP <467> for residual solvents [87] | USEPA Method 524.2 for drinking water [48] |
Case Study: Static Headspace GC-MS for USP <467> Solvents
Experimental Objectives and Workflow
The goal of this experimental work was to develop a single static headspace GC-MS method capable of performing the identification and quantitation of residual solvents as outlined in USP <467>, thereby combining procedures and potentially shortening analysis time [87]. The method aimed to use mass spectrometry to provide superior selectivity compared to the standard Flame Ionization Detection (FID), and to modify system suitability requirements to reduce the use of highly hazardous Class 1 solvents [87].
Diagram: Static Headspace GC-MS Workflow for Residual Solvent Analysis
Research Reagent Solutions
The following table lists the essential materials and reagents used in the development and validation of this static headspace GC-MS method [87].
Table 2: Key Research Reagents and Materials for Static Headspace GC-MS
| Item | Function/Description | Specific Example(s) |
|---|---|---|
| Reference Standards | For identification and calibration of target residual solvents. | USP Class 1 and Class 2 Mixtures (A, B) [87] |
| Sample Diluent | Dissolves the sample matrix without interfering with analysis. | Dimethyl sulfoxide (DMSO), Organic-free water [87] |
| GC Column | Separates volatile compounds based on size and polarity. | DB-624 capillary column (30 m × 0.25 mm, 1.4 µm film) [87] |
| Headspace Vial | Sealed container for sample equilibration. | Standard 22-mL headspace vial with gas-tight septum [87] [48] |
| Carrier Gas | Mobile phase for transporting volatiles through the GC system. | Ultra-high-purity Helium [87] |
| Internal Standard | (Optional) Corrects for analytical variability. | 13C7-toluene (used in study for spiking) [87] |
Detailed Methodology and Instrument Parameters
The experimental work was based on the water-soluble procedure described in USP <467> [87]. Sample preparations involved stepwise dilutions for reference standards and samples, with the final dilution before being placed in the headspace vial being at the residual solvents' allowed concentration limits (the 100% limit concentration) [87].
Finalized Instrument Parameters [87]:
- Headspace Sampler: Vial oven temperature: 80°C; Loop temperature: 90°C; Transfer line temperature: 100°C; Vial equilibration time: 30 min; Pressurization time: 0.5 min.
- Gas Chromatograph: Injector temperature: 140°C; Split ratio: 5:1; Column: DB-624 (30 m x 0.25 mm, 1.4 µm); Oven program: 40°C for 20 min, then ramp to 240°C.
- Mass Spectrometer: Solvent delay: 2.2 min; Scan range: 35-260 amu.
Identification was achieved by combining spectral data (unique quantifying and qualifying ions) with chromatographic retention times. This orthogonal approach allows for the identification of coeluted compounds, which can relax chromatographic resolution requirements and decrease total analysis time [87].
Performance Evaluation and Quantitative Data
The method was validated for its ability to identify and quantify Class 2 residual solvents. A preliminary linearity investigation was conducted at different concentration levels (25% to 200% of the limit concentration) and split ratios (5:1 and 50:1) to establish the dynamic range [87]. The use of MS detection in Selected Ion Monitoring (SIM) mode provided the high sensitivity and improved signal-to-noise ratios necessary for quantifying low-level analytes [87] [88].
Table 3: Example Residual Solvents with Concentration Limits and Associated Ions
| Residual Solvent | USP Class | Concentration Limit (ppm) | Quantifying/Qualifying Ions (m/z) |
|---|---|---|---|
| Benzene | 1 | 2 | 78, 77, 52 |
| Chloroform | 2 | 60 | 118, 83, 85 |
| Methanol | 2 | 3000 | 32, 31, 29 |
| Acetonitrile | 2 | 410 | 41, 40, 39 |
| Hexane | 2 | 290 | 57, 56, 43 |
Note: The ions and concentration limits are illustrative examples based on the cited literature [87].
A recovery study using hydroxyzine dihydrochloride as a water-soluble active pharmaceutical ingredient (API) demonstrated the method's applicability to real-world samples. The study showed that the technique could reliably recover residual solvents like acetonitrile and benzene that were spiked into solutions containing the API [87].
Discussion: Advantages and Challenges of the Static Headspace GC-MS Approach
Key Advantages in a Regulated Environment
The primary advantage of this GC-MS approach is the combination of identification and quantitation into a single procedure, which streamlines the analytical workflow and reduces total analysis time compared to the traditional GC-FID methods that may require multiple instrumental methods and columns [87]. The selectivity of mass spectrometry is a significant improvement, as unique mass spectra can eliminate the need for complete chromatographic resolution of all peaks, allowing for faster GC run times and more confident identification of co-eluting compounds [87].
Furthermore, the method presents a viable path to enhancing laboratory safety. It modifies system suitability requirements, potentially reducing or eliminating the need for the most hazardous Class 1 solvents in daily instrument performance checks [87].
Limitations and Considerations
Despite its advantages, the static headspace GC-MS method presents challenges in quantifying residual solvents at concentrations significantly below their prescribed limits [87]. While static headspace is simple and robust, it can be less sensitive than dynamic headspace techniques, which employ a trapping step to concentrate analytes [12] [86]. This makes dynamic headspace more suitable for applications requiring parts-per-trillion level detection.
The method development process also highlighted that equilibration conditions are critical. Parameters such as temperature, equilibration time, and vial pressure must be carefully optimized, as the concentration of an analyte in the headspace is highly dependent on its partition coefficient (K) between the sample and gas phases [48]. For analytes with high solubility in the sample matrix (high K), even small temperature fluctuations can lead to significant changes in measured peak areas [48].
This case study demonstrates that static headspace GC-MS is a powerful and viable technique for the simultaneous identification and quantitation of Class 1 and Class 2 residual solvents, aligning with the requirements of USP <467>. The method leverages the superior selectivity of MS detection to streamline analysis and improve confidence in results. While the inherent limitations of static headspace, particularly regarding sensitivity for trace-level analysis, make dynamic headspace a better choice for certain applications (e.g., environmental trace analysis), static headspace remains the standard for pharmaceutical residual solvents due to its simplicity, robustness, and direct adoption in regulatory methods. This work underscores the importance of selecting the appropriate headspace sampling technique based on the specific analytical needs, sample matrix, and regulatory context within drug development.
Conclusion
The choice between static and dynamic headspace is not a matter of one being superior to the other, but rather of selecting the right tool for the specific analytical question. Static headspace offers simplicity, robustness, and direct compliance with pharmacopeial methods for routine residual solvent testing, making it a staple in pharmaceutical quality control. Dynamic headspace provides unparalleled sensitivity for trace-level analysis and is indispensable for challenging matrices. The ongoing evolution of these techniques, including greater automation, integration with mass spectrometry, and the development of advanced methods like FET and MVM, promises to further expand their capabilities. For biomedical and clinical research, these advancements will enable more precise profiling of volatile metabolites, improved drug safety profiling, and faster development of innovative therapies, solidifying headspace analysis as a cornerstone of modern analytical science.
References
- 1. The Value of Headspace GC in Volatile Compound Analysis [https://www.pgeneral.com/applications/achieving-accurate-volatile-analysis-the-why-what-and-how-of-headspace-gc/]
- 2. Static or Dynamic Headspace Analysis ? [https://scioninstruments.com/blog/static-or-dynamic-headspace-analysis/]
- 3. Headspace-based approaches for volatile analysis: A review [https://www.koreascience.kr/article/JAKO202513550406322.page]
- 4. Volatile sampling by headspace techniques [https://www.sciencedirect.com/science/article/abs/pii/S0165993615001612]
- 5. Extraction of Ignitable Liquid Residues by Dynamic Capillary... | NIST [https://www.nist.gov/publications/extraction-ignitable-liquid-residues-dynamic-capillary-headspace-sampling-and]
- 6. Finding Ancient Smell: A Comparative Evaluation of Two Headspace ... [https://papers.ssrn.com/sol3/papers.cfm?abstract_id=5222163]
- 7. Optimization of headspace extraction conditions for volatile ... [https://pubs.rsc.org/en/content/articlehtml/2025/ay/d5ay01147g]
- 8. Green Sampling of Gas-Phase Volatile Organic ... [https://www.sciencedirect.com/science/article/pii/S2772577425000989]
- 9. Top Headspace Companies & How to Samplers Them ( Compare ) 2025 [https://www.linkedin.com/pulse/top-headspace-samplers-companies-how-compare-them-2025-fo9cf]
- 10. From Gas to Gas: Fundamentals of Static Headspace ... [https://www.chromatographyonline.com/view/from-gas-to-gas-fundamentals-of-static-headspace-extraction-gas-chromatography]
- 11. Headspace Sampling Fundamentals [https://www.agilent.com/en/product/gas-chromatography/gc-sample-preparation-introduction/what-is-headspace?srsltid=AfmBOopYNnrzaqowP1SZs9LTBzdHPHqb815pu6igRFs57QBtE4xM_45B]
- 12. Static vs. Dynamic Headspace GC: Key Differences Explained [https://www.hplcvials.com/knowledge/static-vs-dynamic-headspace-gc-key-differences-explained.html]
- 13. Troubleshooting Common Issues in Headspace Sampling ... [https://www.pgeneral.com/news/troubleshooting-common-issues-in-headspace-sampling-for-gas-chromatography-2/]
- 14. The LCGC Blog: Thinking Out of the Box on Headspace ... [https://www.chromatographyonline.com/view/lcgc-blog-thinking-out-box-headspace-sampling-gas-chromatography]
- 15. Choosing between static and dynamic headspace methods ... [https://www.linkedin.com/posts/scion-instruments_gaschromatography-headspaceanalysis-analyticalchemistry-activity-7351582753538068482-axVH]
- 16. Systematic comparison of static and dynamic headspace ... [https://pubmed.ncbi.nlm.nih.gov/27526093/]
- 17. Purge and Trap Overview [https://www.teledynelabs.com/products/chromatography/gc-prep/purge-and-trap]
- 18. Dynamic Headspace vs. Purge and Trap: A Comparative Insight [https://www.gerstelus.com/dynamic-headspace-vs-purge-and-trap-a-comparative-insight/]
- 19. Evaluation of dynamic headspace and purge-and-trap ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967399004239]
- 20. 一种利用顶空‑气质联用仪测定挥发有机物的方法 [https://patents.google.com/patent/CN106841469A/zh]
- 21. Headspace Sampling | LCGC International [https://www.chromatographyonline.com/view/headspace-sampling-0]
- 22. Headspace Sampling Fundamentals [https://www.agilent.com/en/product/gas-chromatography/gc-sample-preparation-introduction/what-is-headspace?srsltid=AfmBOorOlqxFY1XMv73Hf1EQ27zfeadDIhDGjeMwJnPR87mJCQ0OMyLu]
- 23. A Generic Headspace GC Method for Residual Solvents in ... [https://www.chromatographyonline.com/view/generic-headspace-gc-method-residual-solvents-pharmaceutical-benefits-rationale-and-adaptations-new]
- 24. Optimizing Sample Introduction for Headspace GC [https://www.chromatographyonline.com/view/optimizing-sample-introduction-headspace-gc]
- 25. Development and validation of headspace gas ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11737362/]
- 26. Headspace Sampling Optimization [https://scioninstruments.com/us/blog/headspace-sampling-optimization/]
- 27. Headspace Sampling: Static vs. Dynamic Techniques [https://www.linkedin.com/posts/scion-instruments_headspace-sampling-static-vs-dynamicwhich-activity-7378402481782878208-I_Mb]
- 28. Full evaporation headspace gas chromatography for ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967313015252]
- 29. Analytical Method by Headspace‐Gas Chromatography for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12614197/]
- 30. analysis of residual solvents using GC-FID with USP ... 467 headspace [https://appslab.thermofisher.com/App/1997/usp-467-analysis-residual-solvents-using-gcfid-with-headspace]
- 31. SW-846 Test Method 5021A: Volatile Organic Compounds ... [https://www.epa.gov/hw-sw846/sw-846-test-method-5021a-volatile-organic-compounds-vocs-various-sample-matrices-using]
- 32. Referred Air Method 25E: Determination of a Vapor Phase ... [https://www.epa.gov/hw-sw846/referred-air-method-25e-determination-vapor-phase-organic-concentration-waste-samples]
- 33. Detection Limits of EPA Method 8260 with Static and ... [https://www.chromatographyonline.com/view/detection-limits-epa-method-8260-static-and-dynamic-headspace-gc-ms-analysis]
- 34. Static Headspace Sampler Unlocking Growth Potential [https://www.archivemarketresearch.com/reports/static-headspace-sampler-220815]
- 35. Experimental Considerations in Headspace Gas ... [https://www.pharmtech.com/view/experimental-considerations-headspace-gas-chromatography]
- 36. Multivariate Optimization Procedure for Dynamic ... [https://www.chromatographyonline.com/view/multivariate-optimization-procedure-for-dynamic-headspace-extractions-coupled-to-gc-gc-]
- 37. Static headspace gas chromatographic method for ... [https://pubmed.ncbi.nlm.nih.gov/15584239/]
- 38. Headspace Samplers Market Report | Global Forecast ... [https://dataintelo.com/report/global-headspace-samplers-market]
- 39. A Fast, Simplified Approach to Quantitative Headspace ... [https://www.chromatographyonline.com/view/fast-simplified-quantitative-headspace-analysis-volatile-impurities-drug-packaging-products]
- 40. Rapid screening of nitrosamine impurities in metformin ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967325007228]
- 41. Headspace Sampling Supplies Competitive Strategies [https://www.marketreportanalytics.com/reports/headspace-sampling-supplies-288839]
- 42. Headspace Sampling Fundamentals [https://www.agilent.com/en/product/gas-chromatography/gc-sample-preparation-introduction/what-is-headspace?srsltid=AfmBOoqN00ZZ3rAs4sq3mfEbfLOKjEJgBgL_VbeRIjfEY7tjOm2sBS9R]
- 43. Thinking Outside The Box On Headspace Sampling For ... [https://www.elementlabsolutions.com/uk/chromatography-blog/post/thinking-outside-the-box-on-headspac]
- 44. The Why, What, and How of Headspace GC [https://www.chromatographyonline.com/view/why-what-and-how-headspace-gc]
- 45. Sample Preparation for Food Contaminant Analysis [https://www.chromatographyonline.com/view/sample-preparation-food-contaminant-analysis]
- 46. Headspace Extraction - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/headspace-extraction]
- 47. A Comparison of GC-FID and SIFT-MS Performance [https://www.mdpi.com/2673-9623/3/2/18]
- 48. Headspace Sampling | LCGC International [https://www.chromatographyonline.com/view/headspace-sampling]
- 49. Comparison of volatile trapping techniques for the ... [https://www.sciencedirect.com/science/article/pii/S0021967320304556]
- 50. Comparative efficiency of soxhlet and accelerated solvent ... [https://www.sciencedirect.com/science/article/pii/S2772577425000242]
- 51. Evaluation of lifetime and analytical performance of gas ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967312004311]
- 52. Evaluation of performance and application validation ... [https://www.sciopen.com/article/10.16791/j.cnki.sjg.2025.08.003]
- 53. Application of static headspace GC–MS for detection ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996924013620]
- 54. The Utility of Headspace Grade Solvents for the Analysis of Organic... [https://www.sigmaaldrich.com/RU/en/technical-documents/technical-article/analytical-chemistry/gas-chromatography/the-utility-of-headspace]
- 55. High Analytical Performance for Untargeted Aroma ... [https://www.chromatographyonline.com/view/high-analytical-performance-for-untargeted-aroma-profiling-by-combining-dynamic-headspace-sampling-capillary-gas-chromatography-and-time-of-flight-mass-spectrometry]
- 56. of Comparison solid-phase microextraction, headspace ... headspace [https://pubmed.ncbi.nlm.nih.gov/22069217/]
- 57. Headspace Gas Chromatography: Types and Uses [https://www.phenomenex.com/knowledge-center/gc-knowledge-center/headspace-gc-principles-applications?srsltid=AfmBOoq0lY57xov2qVNMqJuAflyCUs2wrHfZVGUTcEiKtO_ZkvbFsKHb]
- 58. Headspace Sampling Fundamentals [https://www.agilent.com/en/product/gas-chromatography/gc-sample-preparation-introduction/what-is-headspace?srsltid=AfmBOootAMvBuBj7mkqcyShVthkRiHKQl0ME1bVCvmXZQ3tY_jw5K2ET]
- 59. Optimization of a static headspace GC-MS method and its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9772436/]
- 60. A generic static headspace gas chromatography method ... [https://www.sciencedirect.com/science/article/abs/pii/S002196731001068X]
- 61. Optimization and comparison of headspace hot injection ... [https://www.sciencedirect.com/science/article/abs/pii/S0023643820311440]
- 62. Development and performance evaluation of a novel ... [https://www.sciencedirect.com/science/article/pii/S0021967319305229]
- 63. Optimization of a Dynamic Headspace – Thermal ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914011003328]
- 64. Multiple responses optimization in the development of a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5762238/]
- 65. The Utility of Headspace Grade Solvents for the Analysis ... [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/analytical-chemistry/gas-chromatography/the-utility-of-headspace?srsltid=AfmBOorwLU44Z9UxHdjnxZENF714qSxbq8t2ydfe7h4Y12ffzejq1GvM]
- 66. Is Headspace Sampling Quantitative? [https://www.chromatographyonline.com/view/headspace-sampling-quantitative]
- 67. Enhancing Extractions by Salting Out | LCGC International [https://www.chromatographyonline.com/view/enhancing-extractions-by-salting-out]
- 68. Salting-out assisted liquid–liquid extraction for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6661818/]
- 69. Salting-out Assisted Liquid-Liquid Extraction for the rapid ... [https://www.sciencedirect.com/science/article/abs/pii/S0889157522000758]
- 70. Mobile Phase Selection Guide: Optimal Solvent Pairing ... [https://www.alwsci.com/news/mobile-phase-selection-guide-optimal-solvent-77320943.html]
- 71. Dynamic headspace with ITEX DHS extends ... - PAL System [https://www.palsystem.com/en/sample-prep-injection/micromethods/static-dynamic-headspace/]
- 72. Application of Static Headspace GC-MS Method for ... [https://www.mdpi.com/2304-8158/12/17/3299]
- 73. Solvent Extraction of PDMS Tubing as a New Method for ... [https://link.springer.com/article/10.1007/s10886-024-01469-y]
- 74. Platform headspace gas chromatography method for high ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708523001188]
- 75. Residual Solvent Testing in United States: USP & FDA ... [https://resolvemass.ca/residual-solvent-testing-in-united-states-usp-fda-compliance/]
- 76. Optimized synthesis of suvorexant and determination of eight ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra06779k]
- 77. Development and Validation of a Headspace Gas... | Research Square [https://www.researchsquare.com/article/rs-7203829/v1]
- 78. of Analysis in Pharmaceutical Products... Residual Solvents [https://www.shimadzu.com/an/apl/12763/index.html]
- 79. Residual Solvents by HT3™ Headspace in Reference to ... [https://www.chromatographyonline.com/view/residual-solvents-ht3-headspace-reference-usp-comparison-static-versus-dynamic-headspace-analysis]
- 80. High-Throughput Headspace Analysis of Volatile Impurities ... [https://www.chromatographyonline.com/view/high-throughput-headspace-analysis-of-volatile-impurities-in-consumer-products-using-standard-additions]
- 81. Automated headspace solid-phase dynamic extraction to ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967303018570]
- 82. New Sample Preparation Products and Accessories for ... [https://www.chromatographyonline.com/view/new-sample-preparation-products-and-accessories-for-2024-2025]
- 83. FAQs: <467> Residual Solvents [https://www.usp.org/frequently-asked-questions/residual-solvents]
- 84. ICH Q8 (R2) Pharmaceutical development - Scientific guideline [https://www.ema.europa.eu/en/ich-q8-r2-pharmaceutical-development-scientific-guideline]
- 85. Q8(R2) Pharmaceutical Development November 2009 [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q8r2-pharmaceutical-development]
- 86. Headspace Gas Chromatography - an overview [https://www.sciencedirect.com/topics/chemistry/headspace-gas-chromatography]
- 87. Static Headspace GC–MS Detection of Residual Solvents ... [https://www.chromatographyonline.com/view/static-headspace-gc-ms-detection-residual-solvents-possible-simultaneous-identification-and-quantita]
- 88. A laboratory high‐throughput glass chamber using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6099401/]