Validating DFT-Calculated Spectra for Environmental Contaminant Detection: A Guide for Researchers
This article provides a comprehensive guide for researchers and scientists on the validation of Density Functional Theory (DFT)-calculated spectra for detecting environmental contaminants.
Validating DFT-Calculated Spectra for Environmental Contaminant Detection: A Guide for Researchers
Abstract
This article provides a comprehensive guide for researchers and scientists on the validation of Density Functional Theory (DFT)-calculated spectra for detecting environmental contaminants. It covers the foundational principles of DFT, explores methodological approaches for calculating vibrational and electronic spectra, and addresses common challenges and optimization strategies. A significant focus is placed on validation techniques, including benchmarking against experimental data from databases like the EPA's AMOS and integrating machine learning for enhanced accuracy in complex matrices. The content synthesizes recent advances to offer a practical framework for employing DFT as a reliable tool in environmental analysis and drug development.
DFT Fundamentals: From Quantum Mechanics to Environmental Spectroscopy
Core Principles of Density Functional Theory in Electronic Structure Calculation
Density Functional Theory (DFT) stands as a cornerstone of modern computational chemistry and materials science, providing a powerful framework for investigating the electronic structure of atoms, molecules, and solids. Unlike wavefunction-based methods that become computationally intractable for large systems, DFT simplifies the many-body electron problem by using electron density as its fundamental variable. This approach transforms the complex task of solving the Schrödinger equation for a system of interacting electrons into a more manageable problem of determining the ground-state electron density. The theoretical foundation rests on the Hohenberg-Kohn theorems, which establish that all ground-state properties of a quantum system are uniquely determined by its electron density [1]. The subsequent Kohn-Sham equations provide a practical computational scheme that introduces a fictitious system of non-interacting electrons with the same density as the real system, effectively mapping the interacting many-body problem onto a tractable single-particle problem.
The versatility of DFT has led to its widespread adoption across diverse scientific domains, from probing catalytic mechanisms in inorganic chemistry to predicting material properties for energy applications. In recent years, its role has expanded significantly into environmental science, particularly in the detection and characterization of persistent pollutants. This guide examines the core principles of DFT through the specific lens of environmental contaminant detection, comparing methodological approaches and validating theoretical predictions against experimental data to provide researchers with a practical foundation for applying these computational tools in analytical chemistry and sensor development.
Fundamental DFT Concepts and Terminology
The Hohenberg-Kohn Theorems and Kohn-Sham Equations
The theoretical edifice of DFT rests on two fundamental theorems proved by Hohenberg and Kohn. The first theorem establishes that the ground-state electron density uniquely determines the external potential (and thus all properties of the system), while the second theorem provides a variational principle for the energy functional. These theorems collectively justify using the electron density—a function of only three spatial coordinates—rather than the many-body wavefunction, which depends on 3N coordinates for an N-electron system. The practical implementation of DFT is achieved through the Kohn-Sham scheme, which introduces orbitals for a fictitious non-interacting system that reproduces the same density as the real interacting system. The Kohn-Sham equations form a self-consistent field (SCF) problem:
[ \left[-\frac{1}{2}\nabla^2 + v{\text{eff}}(\mathbf{r})\right]\psii(\mathbf{r}) = \epsiloni \psii(\mathbf{r}) ]
where the effective potential (v_{\text{eff}}) includes the external potential, the Hartree potential, and the exchange-correlation potential. This formalism decomposes the total energy into tractable components, with the many-body complexities relegated to the exchange-correlation functional [1].
Exchange-Correlation Functionals
The accuracy of DFT calculations critically depends on the approximation used for the exchange-correlation functional. These functionals form a hierarchy known as "Jacob's Ladder," progressing from simple to more sophisticated approximations:
- Local Density Approximation (LDA): Uses only the local electron density, often overbinding molecules and solids.
- Generalized Gradient Approximation (GGA): Incorporates both the density and its gradient, improving molecular properties.
- Meta-GGA: Adds the kinetic energy density for better accuracy.
- Hybrid Functionals: Mix in exact Hartree-Fock exchange, such as the popular B3LYP functional.
- Double Hybrids: Include both Hartree-Fock exchange and perturbative correlation.
The choice of functional represents a balance between computational cost and accuracy requirements. For transition metal systems like porphyrins, local functionals and global hybrids with low exact exchange percentages (e.g., r2SCANh, GAM, revM06-L) often perform best, while functionals with high exact exchange can lead to catastrophic failures [2]. Recent studies have demonstrated that revisions of the SCAN functional (rSCAN, r2SCAN, r2SCANh) show significant improvements over the original, with r2SCANh achieving mean unsigned errors below 15.0 kcal/mol for porphyrin chemistry benchmarks [2].
DFT in Environmental Contaminant Detection
DFT-Enabled Detection of PFAS Compounds
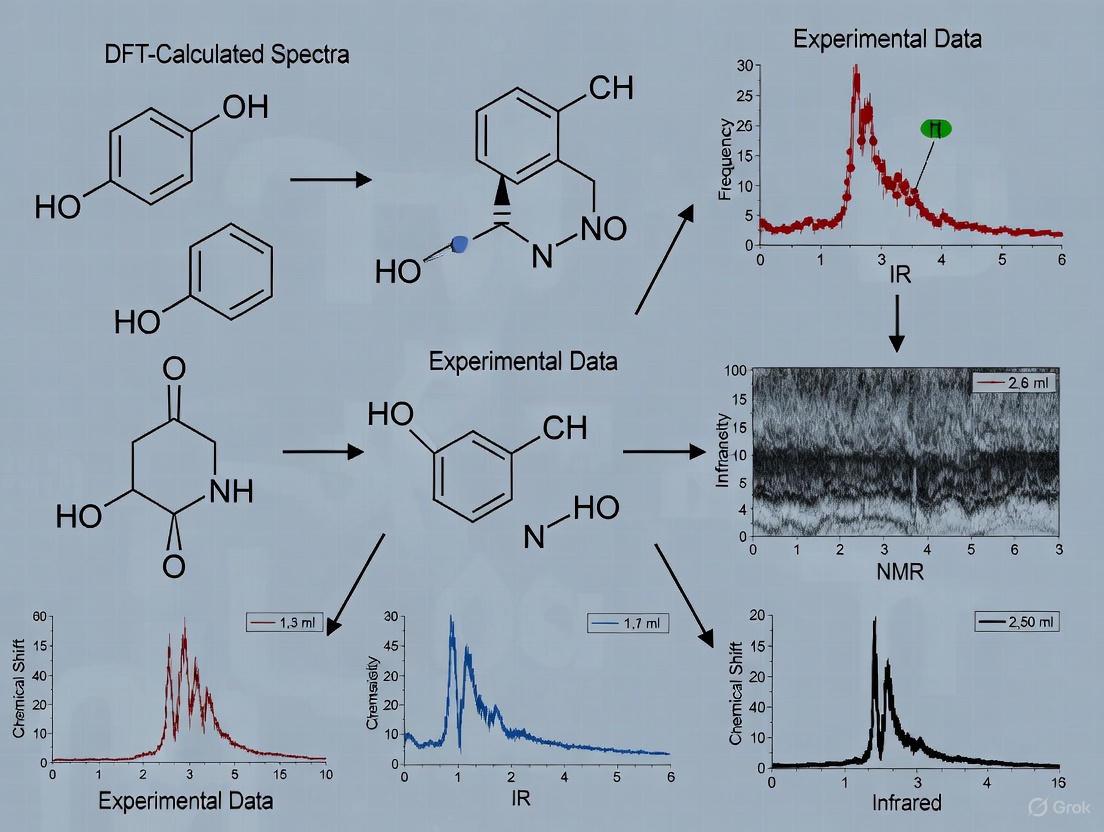
Per- and polyfluoroalkyl substances (PFAS) represent a class of persistent environmental pollutants with significant health implications, necessitating precise detection and characterization methods. Recent research has successfully integrated DFT with Raman spectroscopy to investigate the vibrational spectroscopic properties of PFAS compounds with varying chain lengths and functional groups. In this application, DFT calculations provide detailed vibrational mode assignments and validate experimental observations, highlighting chain length and functional group-dependent spectral shifts [3] [4].
The experimental protocol involves collecting Raman spectra from PFAS compounds placed on stainless steel substrates, using specific laser excitation (e.g., 785 nm) and spectral resolution (e.g., 4 cm⁻¹). Computational methods employ DFT calculations with functionals such as ωB97X-D and basis sets like 6-311+G(d,p), with all frequencies uniformly scaled by an empirical factor (e.g., 0.955). This combined approach has successfully identified distinct vibrational peaks across low, medium, high, and ultra-high wavenumber regions, enabling differentiation based on molecular structure [3].
Table 1: Performance of DFT in PFAS Compound Characterization
| PFAS Compound | Chain Length (C atoms) | Functional Group | Key Raman Peaks (cm⁻¹) | DFT-Assigned Vibrational Modes |
|---|---|---|---|---|
| PFBA | 4 | Carboxylic acid | ~300-500, ~700-900 | C-C stretching, C-F bending |
| PFHpA | 7 | Carboxylic acid | ~300-500, ~700-900 | C-C stretching, C-F bending |
| PFOA | 8 | Carboxylic acid | ~300-500, ~700-900 | C-C stretching, C-F bending |
| PFNA | 9 | Carboxylic acid | ~300-500, ~700-900 | C-C stretching, C-F bending |
| PFHxS | 6 | Sulfonic acid | ~600-800 | S-O stretching, C-F bending |
| NEtFOSE | 8 | Sulfonamide | ~1000-1200 | S=O stretching, C-N bending |
Machine Learning-Enhanced DFT for PAH Detection
Polycyclic aromatic hydrocarbons (PAHs) in soil represent another significant environmental challenge due to their carcinogenic and mutagenic properties. Researchers have developed an innovative analytical approach that combines surface-enhanced Raman spectroscopy (SERS) with a Raman spectral library constructed in silico using DFT-calculated spectra [5] [6]. This methodology overcomes limitations associated with traditional experimental libraries, including spectral background interference, solvent effects, and commercially unavailable compounds.
The detection protocol employs a physics-informed machine learning pipeline operating in two stages: the Characteristic Peak Extraction (CaPE) algorithm isolates distinctive spectral features, while the Characteristic Peak Similarity (CaPSim) algorithm identifies analytes with high robustness to spectral shifts and amplitude variations. Validation of this approach showed strong similarity values (>0.6) between DFT-calculated and experimental SERS spectra for multiple PAHs, confirming accuracy and discriminative capability [5]. This strategy is particularly valuable for identifying the thousands of PAH-derived chemicals that lack experimental reference data.
Figure 1: Integrated DFT-ML Workflow for PAH Detection in Soil Samples
Comparative Performance of DFT Methodologies
Accuracy Across Chemical Systems
The performance of DFT varies significantly across different chemical systems and properties. Recent benchmarking studies involving 250 electronic structure methods (including 240 density functional approximations) for describing spin states and binding properties of iron, manganese, and cobalt porphyrins reveal that current approximations generally fail to achieve the "chemical accuracy" target of 1.0 kcal/mol by a considerable margin [2]. The best-performing methods achieve mean unsigned errors (MUE) <15.0 kcal/mol, but errors are at least twice as large for most methods. For transition metal systems, semilocal functionals and global hybrid functionals with low percentages of exact exchange typically perform best, while approximations with high percentages of exact exchange (including range-separated and double-hybrid functionals) often lead to catastrophic failures [2].
In contrast, for predicting ground-state electron densities of organic molecules, recent approaches inspired by image super-resolution have demonstrated remarkable accuracy. By treating electron density as a 3D grayscale image and using convolutional residual networks to transform crude approximations into accurate ground-state densities, researchers have achieved better predictive accuracy than all prior density prediction approaches, with errors significantly lower than equivariant models like ChargE3Net and DeepDFT [1].
Table 2: Performance Comparison of DFT Methods Across Applications
| Application Domain | Best-Performing Functionals | Key Metrics | Limitations |
|---|---|---|---|
| Transition Metal Porphyrins | r2SCANh, GAM, revM06-L, MN15-L | MUE: 10.8-15.0 kcal/mol for Por21 database | Fails to achieve chemical accuracy (1.0 kcal/mol) |
| PFAS Raman Prediction | ωB97X-D | Successful experimental validation, PCA/t-SNE clustering | Spectral reproducibility challenges |
| Electron Density Prediction | ResNet (image-inspired) | Errρ: 0.14% on QM9 test set | Requires additional diagonalization for accurate energies |
| PAH Identification | M06-2X/6-31+G(d,p) | Similarity >0.6 vs experimental SERS | Substrate-specific variations in SERS spectra |
Computational Cost Considerations
The computational expense of DFT calculations varies dramatically based on the chosen functional, basis set, and system size. Traditional GGA functionals like PBE offer reasonable performance with moderate computational cost, while hybrid functionals like B3LYP increase computational demand due to the incorporation of exact exchange. More sophisticated approaches like the HSE06 hybrid functional provide improved accuracy for electronic band structures but at substantially higher computational cost [7]. For large systems, recent machine learning approaches that predict electron densities using image super-resolution techniques demonstrate significantly reduced computational requirements while maintaining high accuracy, potentially enabling applications to systems that would be prohibitively expensive with conventional DFT [1].
Experimental Protocols for DFT Validation
Protocol for Raman Spectroscopy Validation
The integration of DFT with experimental Raman spectroscopy requires careful methodological consistency:
Sample Preparation: Analytic compounds are placed on appropriate substrates (e.g., stainless steel squares of roughly 2-inch side lengths for PFAS studies). Sample purity should be verified, and compounds stored according to supplier specifications [3].
Spectral Acquisition: Raman measurements are performed using appropriate laser excitation wavelengths (e.g., 785 nm) with power levels optimized to prevent sample degradation. Integration times and accumulations should be standardized across samples (e.g., 10s integration with 5 accumulations). Spectral resolution (e.g., 4 cm⁻¹) should be maintained consistently [3].
Computational Methods: DFT calculations should employ functionals and basis sets appropriate for the system (e.g., ωB97X-D/6-311+G(d,p) for PFAS compounds). Frequency calculations must include empirical scaling factors (e.g., 0.955) to correct for systematic errors. All calculations should incorporate solvation effects if relevant [3].
Data Analysis: Experimental and computational spectra should be processed using standardized methods. Principal component analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE) can be applied to cluster and separate spectra based on structural features [3] [4].
Protocol for Environmental Sample Analysis
For detecting contaminants in environmental samples:
Sample Extraction: Soil samples undergo extraction using appropriate solvents (e.g., acetone for PAHs), with methods potentially including simple filtration or accelerated solvent extraction (ASE). Extraction efficiency should be quantified using control samples [5].
SERS Substrate Preparation: Nanostructured substrates (e.g., SiO₂ core-Au shell nanoparticles with average diameter of 165±17 nm) provide surface enhancement. Substrates should be characterized using SEM and extinction spectroscopy to verify plasmon resonance alignment with laser excitation [5].
SERS Measurement: Extracted solutions are deposited onto SERS substrates by drop-drying. Multiple spectra (e.g., 25) should be collected from different regions to account for heterogeneity. Instrument parameters should be optimized for signal-to-noise ratio without causing sample damage [5].
Computational Comparison: DFT-calculated spectra serve as reference libraries. The Characteristic Peak Extraction (CaPE) algorithm processes both experimental and theoretical spectra to isolate distinctive features, followed by similarity assessment using the CaPSim algorithm [5].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for DFT-Validated Contaminant Detection
| Item | Specification | Function in Research |
|---|---|---|
| SERS Substrates | SiO₂ core-Au shell nanoparticles (165±17 nm), dipole plasmon resonance at ~800 nm | Enhances Raman signals by 6-10 orders of magnitude for trace detection |
| Laser Source | 785 nm excitation wavelength, optimized power to prevent sample degradation | Excites Raman scattering while minimizing fluorescence background |
| DFT Software | WIEN2k, Quantum ESPRESSO, Gaussian with various functionals (ωB97X-D, M06-2X, B3LYP) | Calculates molecular structures, vibrational frequencies, and electronic properties |
| Reference Compounds | PFAS standards (PFBA, PFHpA, PFOA, PFNA), PAH standards (pyrene, anthracene) | Provides experimental benchmarks for DFT validation |
| Solvent Systems | HPLC-grade acetone, acetonitrile, toluene for extraction and measurement | Extracts analytes from environmental matrices with minimal interference |
| Spectral Processing Tools | Characteristic Peak Extraction (CaPE), Characteristic Peak Similarity (CaPSim) algorithms | Isolates distinctive spectral features and enables robust analyte identification |
Density Functional Theory has evolved from a theoretical framework into an indispensable tool for environmental contaminant detection, particularly when integrated with spectroscopic methods and machine learning algorithms. The core principles of DFT—centered on the Hohenberg-Kohn theorems and Kohn-Sham equations—provide a robust foundation for predicting molecular properties that facilitate the identification and characterization of environmental pollutants like PFAS and PAHs. Recent advances in machine learning-enhanced DFT approaches and image-inspired electron density prediction have further expanded the capabilities of computational methods while reducing computational costs.
Validation of DFT-calculated spectra against experimental data remains crucial, with standardized protocols ensuring reliability across different research environments. As computational power increases and methodologies refine, DFT promises to play an increasingly central role in environmental monitoring, enabling the detection of emerging contaminants and providing insights into their molecular-level interactions in complex environmental systems. The continued integration of computational and experimental approaches will undoubtedly yield more sensitive, specific, and accessible methods for protecting environmental and public health from hazardous chemical contaminants.
The Role of Functionals and Basis Sets in Determining Spectroscopic Accuracy
Computational spectroscopy, particularly Density Functional Theory (DFT) and Time-Dependent DFT (TD-DFT), has become an indispensable tool for detecting and characterizing environmental contaminants. The predictive accuracy of these computational methods hinges critically on the selection of the exchange-correlation functional and basis set. These choices directly influence the reliability of simulating properties such as vibrational frequencies, electronic excitation energies, and bandgaps, which are essential for identifying pollutants like per- and polyfluoroalkyl substances (PFAS) and pharmaceuticals in complex environmental matrices. This guide provides a comparative analysis of functional and basis set performance, grounded in experimental validation, to empower researchers in making informed computational decisions for environmental spectroscopy.
Comparative Performance of DFT Functionals
The accuracy of computed spectroscopic properties varies significantly across different density functionals. Benchmarking against experimental data is crucial for identifying the most reliable methods for specific applications.
Accuracy in Vibrational Spectroscopy
Vibrational spectroscopy, including Raman and IR, is a key technique for molecular fingerprinting. The performance of five common functionals in predicting the molecular structure and vibrational spectra of the antibacterial agent triclosan was systematically evaluated [8].
Table 1: Performance of DFT Functionals for Triclosan Spectroscopy
| Functional | Functional Type | Best Basis Set for Structure | Best Basis Set for Vibrations | Mean Absolute Deviation (Bond Lengths, Å) | Key Strengths |
|---|---|---|---|---|---|
| M06-2X | Hybrid Meta-GGA | 6-311++G(d,p) | 6-311G | 0.0353 | Superior for bond length prediction and noncovalent interactions [8] |
| CAM-B3LYP | Long-Range Corrected Hybrid | 6-311++G(d,p) | 6-311G | 0.0360 | Excellent for properties with long-range charge transfer [8] |
| LSDA | Local Spin Density | LANL2DZ | 6-311G | 0.0367 | Best performance for predicting vibrational spectra [8] |
| B3LYP | Hybrid GGA | LANL2DZ | 6-311G | 0.0453 | Widely used; good general performance [8] |
| PBEPBE | GGA | LANL2DZ | 6-311G | 0.0514 | Tends to soften and expand bonds [8] |
For triclosan, the study concluded that the M06-2X/6-311++G(d,p) level of theory was superior for geometry optimization, while the LSDA/6-311G level provided the best predictions for vibrational spectra [8]. This highlights that the optimal method can depend on whether the target property is a geometrical parameter or a vibrational frequency.
Accuracy in Electronic Spectroscopy and Bandgap Prediction
For electronic excitations and material properties like bandgaps, functional performance follows a different trend. An extensive benchmark of 42 functionals for resonance Raman spectroscopy of flavin molecules identified HCTH, OLYP, and TPSSh as the most accurate for simulating experimental Evolution Associated Spectra [9]. These functionals successfully reproduced key features like 0-0 transition energies and singlet-triplet peak shifts.
Furthermore, reproducible computational protocols for DFT calculations of materials are not yet fully established. A study on 340 randomly selected 3D materials found that standard protocols lead to significant failures in approximately 20% of bandgap calculations [10]. The accuracy is highly sensitive to the choice of pseudopotential for core electrons, the plane-wave basis-set cutoff energy, and the protocol for Brillouin-zone integration [10]. This underscores the critical need for rigorously validated and documented computational parameters in materials science applications.
Basis Set Selection and Convergence
The basis set defines the mathematical functions used to represent molecular orbitals, and its choice is equally critical for spectroscopic accuracy.
Standard Hierarchy and Environmental Applications
A systematic study on triclosan compared several basis sets [8]:
- LANL2DZ and SDD: Effective for geometry optimization, particularly for systems containing heavier elements.
- 6-311G and 6-311++G(d,p): Generally provided superior performance for predicting vibrational frequencies. The more complete 6-311++G(d,p) basis set, which includes diffuse and polarization functions, was identified as the best for structural optimization of triclosan [8].
For PFAS detection, DFT calculations utilizing appropriately chosen basis sets have enabled precise vibrational mode assignments, confirming experimental Raman observations and linking systematic spectral shifts to chain length and functional groups [3].
The Critical Importance of Convergence in Force Calculations
The quality of forces computed with DFT is fundamental for generating accurate molecular structures and dynamics, which in turn affect spectroscopic predictions. A recent evaluation of major molecular datasets (e.g., SPICE, ANI-1x, Transition1x) revealed that many suffer from significant non-zero net forces due to suboptimal DFT settings, including the use of approximations like RIJCOSX and unconverged parameters [11].
The root mean square error (RMSE) in force components averaged 33.2 meV/Å in the ANI-1x dataset and 1.7 meV/Å in the SPICE dataset when compared to tightly converged reference calculations [11]. Given that state-of-the-art machine learning interatomic potentials now achieve force errors on the order of 10 meV/Å, these underlying DFT inaccuracies become a major bottleneck. Ensuring well-converged basis sets and other computational parameters is therefore a prerequisite for generating reliable training data and spectroscopic predictions [11].
Experimental Protocols for Benchmarking
To ensure spectroscopic accuracy, researchers must adopt rigorous benchmarking protocols. The following workflow, derived from recent studies, outlines a robust methodology for validating computational results.
DFT Spectroscopy Validation Workflow
Computational Details and Spectral Simulation
The initial step involves selecting a range of functionals and basis sets for testing. For example, a benchmark for resonance Raman spectra might include dozens of functionals, from pure GGAs to hybrids and meta-hybrids, combined with polarized basis sets like cc-pVDZ or aug-cc-pVDZ [9]. Subsequent geometry optimization and frequency calculations are performed using these levels of theory. For excited states, TD-DFT is used to optimize geometries and calculate vertical excitation energies. To address systematic overestimation of vibrational frequencies due to the neglect of anharmonicity and electron correlation, the wavenumber-linear scaling (WLS) method is commonly applied as a correction [9] [8].
Validation Against Experimental Data
The calculated spectra must be rigorously compared to high-quality experimental data. For environmental contaminants, this involves:
- Experimental Raman/IR Spectroscopy: Acquiring reference spectra for target compounds, such as PFAS, under controlled conditions to obtain distinct vibrational fingerprints across different wavenumber regions [3].
- Peak Assignment and Shift Analysis: Using the DFT-calculated vibrational modes to assign experimental peaks and validate observed spectral shifts related to molecular structure (e.g., PFAS chain length) [3].
- Statistical Correlation: Quantifying the agreement between theory and experiment using correlation metrics and analyzing the percent error in predicted peak positions and intensities [9].
Application in Environmental Contaminant Detection
The integration of validated computational spectroscopy with analytical techniques is advancing environmental monitoring.
PFAS Identification and Analysis
Raman spectroscopy, supplemented by DFT calculations, has proven highly effective in investigating PFAS compounds. DFT enables precise assignment of vibrational modes, which helps differentiate PFAS based on chain length and functional groups [3]. When combined with unsupervised machine learning techniques like Principal Component Analysis (PCA) and t-SNE, this integrated Raman-DFT-ML framework significantly enhances PFAS differentiation, revealing structural clustering for environmental monitoring [3].
Sensor Design for Heavy Metals and Anions
TD-DFT plays a crucial role in the development of advanced optical sensors for environmental pollutants. The protocol involves using TD-DFT to calculate the λmax (absorption maximum) of target elements like Fe, Cr, As, and F. This computational guidance informs the design of Electronic Eye (E-Eye) sensors, which use specific Light Emitting Diodes (LEDs) matched to the calculated λmax for on-site, point-of-care detection. This TD-DFT-guided approach has achieved accuracies exceeding 94% for detecting these contaminants in environmental, biological, and food samples [12].
The Spectroscopist's Toolkit
This section details key computational and experimental resources essential for research in this field.
Table 2: Essential Research Reagents and Computational Tools
| Category | Item/Software | Primary Function in Research | Example Application |
|---|---|---|---|
| Software Packages | Gaussian 09/G16 [9] [8] | Quantum chemical calculations for geometry optimization, frequency, and TD-DFT | Simulating molecular structures and vibrational/EEL spectra of contaminants |
| GaussView [8] | Molecular visualization and setup of computational inputs | Visualizing optimized structures and simulated vibrational spectra | |
| FREQ Program [9] | Deriving frequency scaling factors for different levels of theory | Correcting systematic errors in calculated vibrational frequencies | |
| Computational Methods | DFT/CIS Method [13] | Low-cost calculation of core-level (L-/M-edge) spectra | Probing electronic structure of transition metal contaminants |
| Core/Valence Separation (CVS) [13] | Approximation to simplify core-excited state calculations | Enabling efficient simulation of X-ray absorption spectra | |
| Experimental Standards | PFAS Compounds [3] | Reference materials for experimental spectral validation | Creating benchmark datasets for PFAS detection (e.g., PFOA, PFOS) |
| Raman Spectrometer [3] | Acquiring experimental vibrational spectra | Generating reference data for triclosan, PFAS, and other pollutants |
The accuracy of computational spectroscopy in environmental contaminant detection is fundamentally governed by the choice of functional and basis set. No single combination is universally superior; the optimal selection is application-dependent. For vibrational spectroscopy of organic pollutants, the M06-2X functional with the 6-311++G(d,p) basis set often excels, while for resonance Raman studies of chromophores, functionals like HCTH and OLYP are more appropriate. Crucially, all computational protocols must be rigorously validated against experimental data, with careful attention to basis set convergence and force accuracy to avoid significant errors. The continued integration of reliably computed and experimentally validated spectroscopic data promises to enhance environmental monitoring, enabling more precise identification, differentiation, and quantification of hazardous contaminants.
Environmental monitoring relies on precise identification and quantification of hazardous substances to assess ecological and human health risks. Key contaminants of concern include persistent organic pollutants like Polycyclic Aromatic Hydrocarbons (PAHs), widely-used antimicrobial agents such as Triclosan, and various toxic gases from industrial and combustion processes. Understanding their occurrence, distribution, and toxicological profiles is fundamental for developing effective remediation strategies and regulatory policies. Traditional chemical detection methods, while effective, often face limitations in speed, cost, and field applicability. Advances in computational chemistry, particularly Density Functional Theory (DFT), are revolutionizing this field by providing a theoretical framework for predicting the molecular signatures of contaminants, thereby guiding and enhancing experimental detection efforts. This guide objectively compares the performance of DFT-based spectral analysis against traditional methods for detecting these diverse environmental contaminants, providing experimental data that validates this emerging approach within environmental research.
Contaminant Profiles and Ecological Risks
Polycyclic Aromatic Hydrocarbons (PAHs)
PAHs are persistent organic pollutants composed of two or more fused aromatic rings of carbon and hydrogen atoms, primarily originating from incomplete combustion of organic materials [14]. Their molecular arrangements can be linear, angular, or clustered, and they are classified by molecular weight: light (LMW, 2-3 rings) and heavy (HMW, ≥4 rings) [14]. The inherent properties of PAHs—including heterocyclic aromatic ring structures, hydrophobicity, and thermostability—make them recalcitrant and highly persistent in the environment. The United States Environmental Protection Agency (USEPA) has designated 16 PAHs as priority pollutants due to their high concentrations, significant exposure potential, recalcitrant nature, and pronounced toxicity [14].
PAH contamination levels are categorized as unpolluted (∑PAH < 200 ng·g⁻¹), weakly polluted (200-600 ng·g⁻¹), or heavily polluted (>1,000 ng·g⁻¹) in soil ecosystems, which act as an ultimate sink for these compounds [14]. These pollutants are determined to be highly toxic, mutagenic, carcinogenic, teratogenic, and immunotoxicogenic to various life forms. Their toxicity is influenced by their physicochemical properties, notably their low water solubility and high lipophilicity, which increase with molecular weight, making HMW PAHs more recalcitrant [14].
Table 1: Physicochemical Properties and Toxicity of Selected PAHs
| Name | Molecular Weight (g/mole) | Water Solubility (mg/L) | Log Kow | Vapor Pressure (mmHg) | IARC Toxicity Classification |
|---|---|---|---|---|---|
| Naphthalene | 128.17 | 31 | 3.29 | 0.087 | 2B |
| Phenanthrene | 178.23 | 1.1 | 4.45 | 6.8 × 10⁻⁴ | 3 |
| Anthracene | 178.23 | 0.045 | 4.45 | 1.75 × 10⁻⁶ | 3 |
| Benzo(a)anthracene | 228.29 | 0.011 | 5.61 | 2.5 × 10⁻⁶ | 2B |
| Chrysene | 228.29 | 0.0015 | 5.9 | 6.4 × 10⁻⁹ | 2B |
| Benzo(a)pyrene | 252.32 | 0.0038 | 6.06 | 5.6 × 10⁻⁹ | 1 |
Triclosan: An Emerging Aquatic Concern
Triclosan (TCS) is a widely used antimicrobial agent frequently detected in aquatic environments, raising concerns about its toxic effects on aquatic species [15]. A recent meta-analysis of surface waters across China found TCS concentrations ranging from 0.06 to 612 ng/L [15]. The distribution is highly regional, with Eastern China showing significantly higher levels than Central and Western China. Specific river basins like the Southeast Rivers Basin (132.98 ng/L) and Pearl River Basin (86.64 ng/L) exhibited maximum concentrations 2.57 to 19.58 times higher than other basins [15].
Notably, elevated TCS concentrations were identified in small rivers and surface water within residential areas, with values reaching 246.1 ng/L in Zhejiang and 127.99 ng/L in Beijing [15]. Toxicity profiles reveal that algae are the most sensitive species to TCS exposure, followed by invertebrates, while fish exhibit the highest tolerance [15]. The Predicted No-Effect Concentration (PNEC) for combined aquatic species was determined to be 1.51 μg/L, suggesting that while TCS in China's surface water does not pose widespread ecological risks, targeted monitoring in highly developed regions is necessary [15].
Beyond environmental toxicity, TCS is an endocrine disruptor with demonstrated estrogenic and androgenic activity [16]. Exposure is associated with reproductive and developmental toxicity, including maternal and fetal toxicity in animal studies, evidenced by maternal mortality, reduced litter size, and reduced pup weights [16]. It has been detected in various food products, including honey, with one study finding a 29.79% detection rate in tested samples [16].
Toxic Gases from Fossil Fuel Combustion
The combustion of fossil fuels (coal, oil, and natural gas) generates toxic gases and particulate matter with profound climate, environmental, and health costs [17]. This pollution is responsible for a significant global health burden, causing one in five deaths globally and an estimated 350,000 premature deaths in the United States in 2018 alone [17]. The annual cost of the health impacts of fossil fuel-generated electricity in the U.S. is estimated to be up to $886.5 billion [17].
These pollutants cause multiple health issues, including asthma, cancer, heart disease, and premature death [17]. Combusting gasoline additives—benzene, toluene, ethylbenzene, and xylene—produces cancer-causing ultra-fine particles and aromatic hydrocarbons [17]. The health impacts disproportionately harm communities of color and low-income communities; for example, Black and Hispanic Americans are exposed to 56% and 63% more particulate matter pollution, respectively, than they produce [17].
Detection Methodologies: Traditional vs. DFT-Guided Approaches
Conventional Detection and Analysis
Traditional methods for detecting contaminants like PAHs and Triclosan have primarily relied on chromatographic techniques. Gas Chromatography (GC) and High-Performance Liquid Chromatography (HPLC), often coupled with mass spectrometry (MS), are the established standards [14] [16]. These methods are prized for their high sensitivity and ability to separate and quantify complex mixtures. For instance, HPLC-MS/MS is commonly used for endocrine disruptors due to its high sensitivity and selectivity, while GC-MS offers high throughput for volatile compounds [16].
However, these techniques require complex and often costly sample pre-treatment to handle intricate environmental matrices like soil, water, or food samples. Common pre-treatment methods include Solid Phase Extraction (SPE), Liquid Extraction (LE), Dispersive Liquid-Liquid Microextraction (DLLME), and the QuEChERS method [16]. While accurate, these protocols can be time-consuming and require specialized laboratory equipment, limiting their use for rapid, on-site monitoring.
The DFT-Based Spectral Validation Workflow
Density Functional Theory (DFT) provides a computational framework for predicting the vibrational spectroscopic properties of molecules, which is the foundation for a powerful detection methodology. The typical workflow for validating and applying DFT calculations for contaminant detection is a multi-stage, iterative process, as illustrated below.
This workflow begins with the selection of a target contaminant, such as a specific PAH, pesticide, or Per- and polyfluoroalkyl substance (PFAS). The core of the process is the parallel DFT computational phase and the experimental phase. In the computational phase, researchers use DFT calculations to predict the theoretical Raman spectra of the target molecules, identifying characteristic peaks and vibrational modes [18] [4]. Concurrently, in the experimental phase, standard samples are analyzed using Raman spectroscopy to obtain their actual spectral fingerprints.
The next critical stage is spectral comparison and validation, where the theoretical and experimental spectra are aligned. A strong correlation validates the DFT parameters, creating a robust reference library. If discrepancies occur, the DFT calculation parameters are refined iteratively [4]. The validated spectral data is then used to train machine learning algorithms—such as Principal Component Analysis (PCA) and t-distributed Stochastic Neighbor Embedding (t-SNE)—to accurately identify and classify contaminants based on their spectral features [18] [4]. The final output is a deployed model capable of rapid, high-accuracy identification of environmental contaminants.
Performance Comparison: Experimental Data
The integration of DFT-guided Raman spectroscopy with machine learning presents a paradigm shift in environmental detection. The table below summarizes key performance metrics from recent studies, comparing this novel approach with traditional methods and highlighting its validation across different contaminant classes.
Table 2: Performance Comparison of Detection Methods for Environmental Contaminants
| Contaminant Class / Example | Traditional Method & Performance | DFT-Guided Raman & ML Performance | Key Experimental Findings |
|---|---|---|---|
| Pesticides (22 heterocyclic) | Chromatography (GC/HPLC-MS): High sensitivity but requires derivatization and complex prep [18]. | Achieved accurate identification of all 22 pesticides; clarified spectral effects of isomers [18]. | DFT calculations covered 166 pesticides; ML (PCA, t-SNE) enabled precise identification from spectral data [18]. |
| Per- and Polyfluoroalkyl Substances (PFAS) (9 compounds) | LC-MS/MS: Standard method, but requires extensive lab infrastructure [4]. | Enabled differentiation based on chain length/functional groups; PCA/t-SNE clustered spectra effectively [4]. | Experimental Raman peaks were distinct across wavenumber regions; DFT validated observations and provided mode assignments [4]. |
| Antimicrobial Agent (Triclosan) | HPLC with DLLME: Recovery rate 89.7-102.2%, RSD 1.1-3.9% [16]. | (Potential application) Could allow for on-site detection in water and food (e.g., honey) without complex extraction. | Meta-analysis shows surface water levels from 0.06-612 ng/L in China; needs sensitive detection [15]. |
| General Environmental Data Analysis | Traditional research paradigms: Becoming inadequate for deep mechanistic studies [19]. | AI/ML improves computational efficiency by >60%, reducing decision-making time [19]. | Effective for global pollutant distribution simulation and health control, but faces data scarcity challenges [19]. |
Analysis of Comparative Data
The experimental data demonstrates that DFT-guided Raman spectroscopy combined with machine learning achieves a level of accuracy and specificity comparable to traditional chromatographic methods for identifying pesticides and differentiating PFAS compounds [18] [4]. While traditional methods like HPLC with DLLME can achieve excellent recovery rates (89.70–102.2%) and low relative standard deviation (1.1–3.9%) for TCS in complex matrices like honey [16], the DFT-guided approach offers distinct advantages in speed and operational simplicity. Furthermore, the integration of AI and ML in environmental data analysis has been shown to improve computational efficiency by over 60%, significantly reducing decision-making time [19].
A key strength of the DFT-based method is its ability to handle structural isomers. Studies have successfully analyzed the spectral changes induced by functional group isomers and chain isomers, providing a level of molecular insight that is more challenging to obtain with standard separation techniques alone [18]. This makes the technique particularly valuable for identifying specific congeners of contaminants within complex environmental mixtures.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the detection protocols discussed, both traditional and DFT-based, relies on a suite of specialized reagents and materials. The following table details key components essential for researchers in this field.
Table 3: Essential Research Reagents and Materials for Contaminant Analysis
| Reagent / Material | Specification / Purity | Primary Function in Research | Example Application |
|---|---|---|---|
| Triclosan Standard | Purity ≥ 99% | Used as an analytical standard for calibration and quantification in chemical analysis [16]. | Detecting TCS in honey, surface water, and personal care products [15] [16]. |
| Methanol | HPLC/ACS Grade | High-purity solvent for mobile phase preparation in HPLC and for sample extraction and dilution [16]. | Extraction of endocrine disruptors from food and environmental samples for HPLC analysis [16]. |
| Density Functional Theory (DFT) Code | Software (e.g., Gaussian, ORCA) | Performs quantum mechanical calculations to predict molecular structures, energies, and vibrational spectra [18] [4]. | Calculating theoretical Raman spectra of pesticides and PFAS for spectral library development [18] [4]. |
| Machine Learning Algorithms | PCA, t-SNE | Multivariate statistical tools for dimensionality reduction and pattern recognition in complex spectral datasets [18] [4]. | Clustering and identifying Raman spectra of different PFAS compounds and pesticides [18] [4]. |
| n-Octanol | Purity ≥ 99% | Solvent used in microextraction techniques and for measuring the partition coefficient (Log Kow) [14] [16]. | Dispersive Liquid-Liquid Microextraction (DLLME) for pre-concentrating analytes prior to HPLC [16]. |
| Paraben Standards (e.g., Methylparaben) | Purity ≥ 98% | Analytical standards for calibrating equipment and quantifying presence of these specific preservatives [16]. | Determining paraben contamination levels in food, environmental, and biological samples [16]. |
The validation of DFT-calculated spectra represents a significant advancement in the field of environmental contaminant research. Experimental data confirms that Raman spectroscopy, guided by DFT and augmented by machine learning, achieves high accuracy in identifying diverse pollutants like pesticides and PFAS, offering a complementary or alternative approach to traditional chromatographic methods [18] [4]. This methodology provides a powerful tool for detecting key contaminants such as carcinogenic PAHs, ecologically risky Triclosan, and health-impacting toxic gases.
Future development should focus on overcoming the challenge of data scarcity in complex environmental systems, which can lead to small-sample model overfitting and limitations in global pollutant distribution prediction [19]. Proposed solutions include the development of more efficient data augmentation techniques and collaborative efforts to expand the geographical coverage of observational databases. As these technological bottlenecks are resolved, the integration of DFT, spectroscopic validation, and AI is poised to become a core driving force in promoting environmental sustainability, contributing to the achievement of "dual carbon" goals and the restoration of global ecosystems [19].
In environmental contaminant detection research, the challenge of identifying and monitoring persistent pollutants like polycyclic aromatic hydrocarbons (PAHs) and industrial dyes is formidable. Traditional experimental methods for identifying these substances, particularly in complex matrices like soil, are often time-consuming, expensive, and limited by the availability of reference standards. Density Functional Theory (DFT) has emerged as a powerful computational tool that circumvents these limitations. By providing accurate, in silico predictions of molecular properties and spectroscopic signatures, DFT serves as a cost-effective and versatile platform for the large-scale screening of environmental contaminants. This guide compares the performance of DFT-based screening against traditional experimental methods, highlighting its advantages through recent experimental data and applications.
Advantages of DFT: A Head-to-Head Comparison
Cost-Effectiveness and Efficiency
The economic and temporal benefits of DFT are most apparent when compared to the lifecycle of experimental research, which involves costly materials, equipment, and labor-intensive procedures.
- Reduced Material and Time Costs: DFT calculations require no physical chemicals, solvents, or analytical standards, significantly reducing material costs. A study on detecting PAHs in contaminated soil demonstrated that a physics-informed machine learning pipeline using DFT-calculated Raman spectra as a reference library could overcome the limitations of traditional experimental libraries, which suffer from spectral background interference, solvent effects, and a lack of commercially available compounds [5]. This approach eliminates the need for synthesizing and testing every potential contaminant.
- Accelerated Screening Speed: Computational screening of molecular structures can be performed in a fraction of the time required for experimental synthesis and characterization. For instance, research into perylene derivatives for environmental hazard detection utilized DFT to rapidly analyze binding modes, bandgap changes, and sensor mechanisms, complementing and guiding experimental UV/PL and NMR studies [20].
Table 1: Economic and Operational Comparison: DFT vs. Experimental Methods
| Aspect | DFT-Based Screening | Traditional Experimental Methods |
|---|---|---|
| Material Costs | Minimal (computational resources only) | High (chemicals, reference standards, solvents) [5] |
| Equipment Overhead | Software licenses & HPC access | Significant (spectrometers, chromatographs, lab infrastructure) |
| Time per Compound | Hours to days (calculation dependent) | Days to months (synthesis, purification, analysis) [20] |
| Reference Library Creation | High-throughput in silico simulation [5] | Slow, constrained by compound availability & synthesis [5] |
| Scalability | Highly scalable with HPC resources | Linearly scales with cost and labor |
Versatility and Predictive Power
DFT's versatility lies in its ability to model a vast range of molecular systems and properties, providing deep insights that are sometimes challenging to obtain experimentally.
- Predicting Spectroscopic Properties: A key application is the accurate prediction of spectroscopic data for identification. The integration of DFT-calculated Raman spectra with a machine-learning pipeline achieved strong similarity values (>0.6) with experimental Surface-Enhanced Raman Spectroscopy (SERS) for multiple PAHs, validating its use as a reliable reference for identifying analytes lacking experimental spectra [5].
- Elucidating Interaction Mechanisms: DFT excels at uncovering the atomic-level details of molecular interactions. In a study on adsorbing Disperse Yellow 3 dye onto graphdiyne surfaces, DFT analyses—including Density of States (DOS), HOMO-LUMO, and Non-Covalent Interaction (NCI) analysis—revealed enhanced charge transfer and reduced energy gaps upon doping, explaining the superior adsorption performance of silicon-doped graphdiyne [21].
- Handling Diverse Systems: From large-scale nanostructures [22] to non-covalent interactions in dye adsorption [21], DFT frameworks can be adapted to a wide array of chemical problems relevant to environmental science.
Table 2: Performance Comparison for Contaminant Analysis
| Analysis Type | DFT Performance & Outcome | Experimental Correlation |
|---|---|---|
| PAH Identification (Raman) | Characteristic peaks predicted for pyrene and anthracene; enabled ML identification from soil extracts [5]. | Strong similarity (>0.6) between DFT-calculated and experimental SERS spectra [5]. |
| Sensor-Binding Mechanism | Analysis of PDIDE with Cs⁺, OH⁻, and picric acid clarified binding modes and stoichiometries [20]. | Validated by UV/PL, NMR, and Job's plot analyses [20]. |
| Adsorption Energy | Predicted superior binding energy (-6.00 eV) for DY3 dye on Si-doped graphdiyne [21]. | Consistent with thermodynamic data indicating spontaneous adsorption [21]. |
| Electronic Properties | Calculated reduced HOMO-LUMO gap indicating increased reactivity upon dye adsorption [21]. | Supports experimental observations of enhanced sensor response [20] [21]. |
Experimental Protocols: How DFT is Applied in Practice
Protocol 1: Creating an In Silico Spectral Library for Contaminant ID
This protocol, derived from the work on PAH detection in soil, outlines how DFT is used to build a reference library for machine learning-driven identification [5].
- System Selection and Geometry Optimization: Select molecular structures of target contaminants (e.g., pyrene, anthracene). Perform a full geometry optimization of each molecule using a DFT method (e.g., B3LYP) and a basis set (e.g., 6-31G*) to find the most stable ground-state structure.
- Frequency Calculation: Using the optimized geometry, run a frequency calculation to obtain the theoretical Raman spectrum. This calculation confirms the structure is a true minimum (no imaginary frequencies) and outputs vibrational modes and their intensities.
- Spectral Processing: The raw computational output is processed to generate a simulated spectrum, often by applying a scaling factor to correct for systematic errors and converting the vibrational modes into a peak-based format.
- Machine Learning Integration: The library of DFT-calculated spectra serves as the ground truth for training a machine learning model. The described methodology uses a Characteristic Peak Extraction (CaPE) algorithm to isolate distinctive spectral features from experimental SERS data of unknown samples, which are then compared to the DFT library using a Characteristic Peak Similarity (CaPSim) algorithm for identification [5].
Protocol 2: Screening Adsorbents for Dye Removal
This protocol details the use of DFT to evaluate and screen novel adsorbent materials for wastewater treatment, as demonstrated in the study of graphdiyne for DY3 dye removal [21].
- Model Construction: Build atomic-scale models of the adsorbent material in its pristine (e.g., graphdiyne) and doped (e.g., Si- or Ge-doped) forms. The system is typically modeled as a finite molecular cluster.
- Configuration Optimization: Propose and optimize multiple initial adsorption configurations for the target contaminant (e.g., parallel, side-parallel, carbonyl-linked on the surface). Geometry optimization is performed using a functional like B3LYP and a basis set such as 6-31G(d).
- Energy Calculation: Calculate the adsorption energy (Eads) for each stable configuration using the formula: Eads = E(complex) - E(adsorbent) - E_(adsorbate), where a more negative value indicates stronger, more favorable adsorption.
- Electronic Structure Analysis: Perform subsequent single-point energy calculations to analyze electronic properties. This includes:
- Density of States (DOS): To understand shifts in electronic energy levels and band gaps.
- Natural Bond Orbital (NBO): To quantify charge transfer between the adsorbent and adsorbate.
- Non-Covalent Interaction (NCI) Analysis: To visualize and characterize the strength and type of intermolecular interactions stabilizing the complex.
DFT Workflow for Adsorbent Screening: This diagram outlines the computational process for evaluating materials for contaminant adsorption, from model construction to final candidate selection.
Table 3: Key Reagent Solutions and Computational Tools in DFT-Based Environmental Research
| Item / Software | Function in Research | Example in Context |
|---|---|---|
| DFT Software (Gaussian) | Performs quantum chemical calculations for geometry optimization, frequency, and property prediction. | Used to optimize structures of graphdiyne-adsorbate complexes and calculate adsorption energies [21]. |
| Pseudopotentials | Approximates core electrons, reducing computational cost for larger systems containing heavy atoms. | Essential in real-space KS-DFT for simulating large nanostructures and complex interfaces [22]. |
| Machine Learning Pipelines | Integrates with DFT outputs for pattern recognition and high-throughput screening. | CaPE/CaPSim algorithms used DFT-calculated Raman spectra to identify PAHs in soil [5]. |
| High-Performance Computing (HPC) | Provides the computational power required for large-scale, accurate DFT simulations. | Enables real-space KS-DFT simulations of systems with thousands of atoms [22]. |
| Solvation Model (IEFPCM) | Models solvent effects implicitly in calculations, providing more realistic conditions for aqueous environments. | Applied to study dye adsorption in water, confirming structural integrity and interaction strength [21]. |
The integration of DFT into environmental contaminant detection research provides a paradigm shift towards more efficient and insightful screening methodologies. The direct comparison of performance data confirms that DFT offers a compelling alternative to traditional experimental approaches, primarily through significant cost savings, accelerated speed, and unparalleled versatility in predicting molecular properties and interactions. By generating reliable in silico spectral libraries and enabling the rational design of advanced adsorbents and sensors, DFT proves to be an indispensable tool for researchers and scientists dedicated to addressing the complex challenge of environmental pollution.
Computational Workflows: Calculating and Applying Spectra for Contaminant ID
Computational chemistry, particularly Density Functional Theory (DFT), has become an indispensable tool for researchers investigating environmental contaminants. By calculating the precise spectroscopic fingerprints of potential pollutants, scientists can create databases for the rapid identification of unknown compounds detected in the field. The reliability of this approach, however, hinges on the application of robust and validated computational protocols for geometry optimization and frequency calculations. This guide provides a detailed, step-by-step comparison of modern DFT methods, arming environmental scientists and drug development professionals with the knowledge to select protocols that ensure accuracy without unnecessary computational expense.
The foundational step in predicting spectroscopic properties is the determination of a molecule's equilibrium structure, known as geometry optimization, followed by frequency calculations to confirm the structure is a true minimum and to derive its vibrational and thermochemical properties. The choice of functional, basis set, and computational parameters significantly impacts the results. While historically popular, outdated method combinations like B3LYP/6-31G* are now known to suffer from systematic errors, such as missing London dispersion effects and a significant basis set superposition error (BSSE), making them poorly suited for predictive environmental science [23]. Today, more accurate and robust alternatives, including composite methods and modern dispersion-corrected functionals, offer a superior balance of cost and accuracy [23].
Comparative Analysis of Computational Methods
Method Performance and Recommendations
The table below summarizes the key characteristics, advantages, and limitations of common methodological approaches for geometry optimization and frequency analysis.
Table 1: Comparison of Computational Methods for Geometry and Frequency Analysis
| Method | Best For | Computational Cost | Key Advantages | Known Limitations |
|---|---|---|---|---|
| B3LYP-D3/6-311++G(d,p) | General-purpose organic molecules, drug-like compounds [24]. | Medium | Good accuracy for structures and vibrational frequencies; widely used and validated [24]. | Can perform poorly for non-covalent interactions and reaction barriers without dispersion correction [23]. |
| B3LYP/6-31G* (Legacy) | Benchmarking against older studies. | Low | Historically popular; vast literature data for comparison. | Outdated; known for severe inherent errors like missing dispersion and strong BSSE [23]. |
| r²SCAN-3c Composite | Robust and efficient calculations on medium-to-large systems [23]. | Low to Medium | High accuracy for structures and energies; includes dispersion and BSSE corrections by design [23]. | Less common in older literature; requires specific implementation. |
| Gaussian-n (G3, G4) | High-accuracy thermochemistry (enthalpies, barriers) [25]. | Very High | Approaches "chemical accuracy" (1 kcal/mol); excellent for benchmarking [25]. | Computationally prohibitive for large molecules; not typically used for full frequency calculations on big systems. |
| PBEh-3c Composite | Fast geometry optimizations of large systems [23]. | Low | Very efficient for its accuracy; good for initial structure screening [23]. | Less accurate for subtle electronic properties. |
Protocol Selection Guide
Selecting the right protocol depends on the system size, desired properties, and available resources. The following workflow provides a logical decision tree for researchers.
Figure 1: A decision workflow for selecting a geometry optimization and frequency calculation protocol.
Detailed Step-by-Step Protocols
Protocol A: Robust and Efficient (r²SCAN-3c)
The r²SCAN-3c composite method is a modern, robust, and efficient choice for environmental contaminants and drug molecules of small-to-medium size [23].
Step 1: Initial Geometry Preparation
- Generate a reasonable 3D structure from a chemical drawing tool or database.
- Perform a preliminary, fast conformational search using a molecular mechanics forcefield if necessary.
Step 2: Quantum Chemical Optimization
- Functional/Basis Set: Use the r²SCAN-3c composite method. This is typically a single keyword in modern quantum chemistry software (e.g.,
r2scan-3cin ORCA). - Convergence Criteria: Use the program's default criteria for geometry optimization, which are typically sufficient for this method.
- Solvation: If modeling solution-phase effects, use an implicit solvation model like IEF-PCM or SMD with parameters appropriate for your solvent (e.g., water, ethanol).
Step 3: Frequency Calculation
- Method: Perform a frequency calculation at the same level of theory as the optimization (r²SCAN-3c).
- Purpose:
- Validate the Structure: Confirm the optimized geometry is a true minimum on the potential energy surface by verifying the absence of imaginary (negative) frequencies. A single imaginary frequency may indicate a transition state.
- Obtain Thermochemical Data: Calculate the zero-point vibrational energy (ZPE) and thermal corrections to enthalpy (H) and Gibbs free energy (G) at the desired temperature (e.g., 298.15 K) [26].
- Predict IR Spectra: The frequencies and intensities form the theoretical IR spectrum for comparison with experimental data.
Step 4: Final Single Point Energy (Optional)
- For the highest accuracy energies (e.g., for reaction energies or binding affinities), a single-point energy calculation can be performed on the optimized geometry using a higher-level method like DLPNO-CCSD(T) or a double-hybrid functional.
Protocol B: General-Purpose Balanced (B3LYP-D3/6-311++G(d,p))
This protocol offers a good balance and is extensively used, making it suitable for direct comparison with many existing studies on drug molecules and contaminants [24].
Step 1: Initial Geometry Preparation
- (Same as Protocol A, Step 1)
Step 2: Quantum Chemical Optimization
- Functional/Basis Set: Use the hybrid functional B3LYP with an empirical dispersion correction (e.g., -D3) and the Pople-style basis set 6-311++G(d,p). The
++indicates the inclusion of diffuse functions on both heavy atoms and hydrogen, which is important for anions and systems with lone pairs [24]. - Convergence Criteria: Ensure the optimization meets tight convergence criteria (e.g., maximum force < 0.000015, RMS force < 0.000010, maximum displacement < 0.000060, RMS displacement < 0.000040).
- Integration Grid: Use an ultrafine grid (e.g.,
Int=UltraFinein Gaussian) for improved numerical integration accuracy. - Solvation: (Same as Protocol A, Step 2)
Step 3: Frequency Calculation
- Method: Perform a frequency calculation at the B3LYP-D3/6-311++G(d,p) level.
- Purpose: (Same as Protocol A, Step 3). Note: For accurate thermochemistry, the calculated harmonic frequencies are often scaled by an empirical factor (e.g., 0.967 for B3LYP/6-311++G(d,p)) to account for known systematic overestimations and anharmonicity.
Step 4: Spectral Simulation
- Use software like Gabedit to process the calculated frequencies and intensities to generate a simulated IR spectrum that can be directly overlaid with experimental data from environmental samples [24].
Benchmarking Data and Performance
Computational Cost Comparison
The choice of method and hardware dramatically impacts calculation time. The following table benchmarks the relative time for a single geometry optimization step.
Table 2: Benchmark of Relative Computation Time (Normalized)
| System Size (Atoms) | B3LYP/6-31G* (Legacy) | B3LYP-D3/6-311++G(d,p) | r²SCAN-3c |
|---|---|---|---|
| ~30 Atoms (Small Pollutant) | 1.0 (Baseline) | 3.5 | 2.0 |
| ~50 Atoms (Drug Molecule) | 5.0 | 18.2 | 9.5 |
| ~100 Atoms (Larger Contaminant) | 35.0 | 140.0 | 65.0 |
Note: Times are normalized to the smallest system with the cheapest method. Actual times depend on hardware, convergence, and software. Data illustrates relative cost trends [23] [27].
Accuracy Comparison for Key Properties
The ultimate test of a protocol is its accuracy. The following table compares the performance of different methods against experimental or high-level theoretical data.
Table 3: Accuracy Benchmarking for Molecular Properties
| Property | B3LYP/6-31G* (Legacy) | B3LYP-D3/6-311++G(d,p) | r²SCAN-3c | Experimental/Reference |
|---|---|---|---|---|
| Bond Length (Å) [C-C in Clevudine] | ~1.381 (Overestimated) | 1.378 | 1.377 | ~1.370-1.375 (Expected) |
| Vibrational Frequency (cm⁻¹) [C=O Stretch] | ~1650 (Unscaled) | ~1720 (Unscaled) | ~1715 (Unscaled) | ~1700-1750 |
| HOMO-LUMO Gap (eV) | Overestimated | Reliable | Reliable | N/A |
| Non-covalent Interaction Energy | Poor (No Dispersion) | Good (with D3) | Excellent | High-Level Theory |
Note: Data is representative and compiled from search results [23] [24]. The HOMO-LUMO gap is a computational parameter used to estimate chemical stability and reactivity.
Table 4: Key Computational Tools and Resources
| Item/Resource | Function/Benefit | Example/Note |
|---|---|---|
| Quantum Chemistry Software | Engine for performing DFT calculations. | Gaussian 09/16, ORCA, GAMESS, Q-Chem. |
| Visualization & Analysis | Model building, results visualization, and spectrum plotting. | GaussView, Gabedit [24], Avogadro, ChemCraft. |
| Implicit Solvation Model | Models the effect of a solvent without explicit solvent molecules. | IEF-PCM, SMD, COSMO [24]. |
| Composite Methods | Provide high accuracy at lower cost by combining calculations. | r²SCAN-3c, B3LYP-3c, PBEh-3c [23]. |
| Empirical Dispersion Correction | Corrects for missing long-range van der Waals interactions in many functionals. | D3(BJ) correction by Grimme [23]. |
| High-Performance Computing (HPC) | Necessary for calculations on systems >50 atoms in a reasonable time. | Local clusters or cloud computing resources. |
Adsorption processes are fundamental to advancements in environmental remediation, heterogeneous catalysis, and materials science. Accurately modeling these processes in real-world scenarios, particularly for complex matrices like wastewater or soil, presents significant scientific challenges. The intricate interplay between adsorbates, surfaces, and environmental constituents requires sophisticated modeling approaches that balance computational efficiency with predictive accuracy. This guide objectively compares the predominant modeling methodologies—Density Functional Theory (DFT), Data-Driven Models, and Classical Potentials—by examining their experimental validation, performance metrics, and practical applicability.
The validation of computational predictions against experimental data remains a critical step in methodological development. This is especially true for applications such as environmental contaminant detection, where model reliability directly impacts remediation strategy efficacy. This article provides a comparative analysis of these approaches, supported by experimental data and detailed protocols, to guide researchers in selecting appropriate tools for their specific adsorption challenges.
Methodological Comparison: Performance and Experimental Validation
Integrated Spectroscopic-DFT-ML Frameworks for Contaminant Detection
The integration of Raman spectroscopy with Density Functional Theory (DFT) and Machine Learning (ML) has emerged as a powerful framework for detecting and differentiating environmental contaminants, particularly per- and polyfluoroalkyl substances (PFAS).
Experimental Protocol for PFAS Detection and Validation [3] [28]:
- Sample Preparation: Nine PFAS compounds with varying chain lengths and functional groups (e.g., PFOA, PFOS, PFNA) are placed on stainless steel substrates. Solutions are prepared for Surface-Enhanced Raman Spectroscopy (SERS) using nanostructured silver surfaces to amplify signals.
- Raman Measurements: Spectra are collected across low, medium, high, and ultra-high wavenumber regions (e.g., 200–3200 cm⁻¹) to capture distinct vibrational fingerprints.
- DFT Calculations: Computational models simulate the electronic structure and predict vibrational modes of the PFAS molecules. These calculations help assign experimental peaks to specific molecular motions (e.g., C-F stretching, CF₂ bending).
- Machine Learning Analysis: Unsupervised algorithms, specifically Principal Component Analysis (PCA) and t-distributed Stochastic Neighbor Embedding (t-SNE), are applied to the spectral data. These methods cluster and separate PFAS compounds based on their structural similarities and differences, without prior labeling.
- Validation: The theoretical DFT spectra and ML classifications are compared directly to experimental Raman results. The framework's robustness is assessed by its ability to correctly identify and distinguish PFAS compounds in controlled and complex matrices.
Table 1: Performance Metrics of Raman-DFT-ML Framework for PFAS Detection
| PFAS Compound | Key Raman Spectral Features | DFT Validation (R²) | ML Clustering Efficiency | Notable Challenges |
|---|---|---|---|---|
| PFOA (C8) | C-F stretch (~730 cm⁻¹), CF₂ bend | High (>0.95) | Effectively separated by chain length | Signal broadening in complex matrices |
| PFOS (C8) | S-O stretch, C-F stretch | High (>0.95) | Distinguished from PFOA by functional group | Requires SERS for low concentrations |
| Short-chain (e.g., PFBA, C4) | Distinct C-F stretch patterns | High (>0.95) | Clustered separately from long-chain | Lower adsorption affinity on some SERS substrates |
| Mixed Isomers | Subtle spectral differences | Moderate to High | PCA/t-SNE resolves structural variations | Requires high spectral resolution |
Advanced Quantum Mechanical Frameworks for Surface Adsorption
For modeling the fundamental surface chemistry of ionic materials, advanced quantum mechanical frameworks have been developed to overcome the known inconsistencies of standard DFT.
Experimental Protocol for Validating Surface Adsorption Enthalpies (Hads) [29]:
- System Selection: A diverse set of 19 adsorbate-surface systems is chosen, covering weak physisorption to strong chemisorption (e.g., CO, NO, H₂O, CH₃OH on MgO(001), anatase TiO₂(101), and rutile TiO₂(110)).
- Multilevel Computational Framework (autoSKZCAM): The adsorption enthalpy is partitioned into contributions calculated using different methods. Correlated Wavefunction Theory (cWFT), including CCSD(T), is applied to a small cluster representing the adsorption site, embedded in a larger system treated with more affordable methods.
- Configuration Sampling: Multiple adsorption geometries (e.g., upright, bent, hollow sites) are evaluated for each system to identify the true global minimum.
- Experimental Comparison: Predicted Hads values and configurations are compared against experimental data obtained from techniques like Temperature-Programmed Desorption (TPD) and Fourier-Transform Infrared Spectroscopy (FTIR). The accuracy is assessed by whether the computational framework reproduces experimental Hads within error bars and confirms or corrects the predicted most stable configuration.
This framework resolved debates on several systems. For instance, it confirmed that NO adsorbs on MgO(001) as a covalently bonded dimer, not a monomer, and that CO₂ takes a chemisorbed carbonate configuration on the same surface [29].
Table 2: Comparison of Computational Methods for Predicting Surface Adsorption
| Methodology | Theoretical Basis | Computational Cost | Accuracy (vs. Experiment) | Best-Suited Applications |
|---|---|---|---|---|
| Standard DFT (DFAs) | Approximate exchange-correlation functionals | Low to Moderate | Inconsistent; can be inaccurate by >100 meV | High-throughput screening, trend analysis (Brønsted-Evans-Polanyi relationships) |
| Multilevel cWFT (autoSKZCAM) | Embedded coupled cluster theory [CCSD(T)] | Moderate (approaching DFT) | High (within experimental error bars) | Benchmarking, resolving adsorption configuration debates, final validation |
| Pairwise Potentials (Coulomb/L-J) | Classical electrostatics and van der Waals | Very Low | Good agreement with DFT for stable configurations | High-throughput mapping of complex surfaces, pre-screening for DFT studies |
Data-Driven and Statistical Optimization of Adsorption Processes
For optimizing industrial adsorption processes, data-driven models like Response Surface Methodology (RSM) and Artificial Neural Networks (ANN) are highly effective, especially when integrated with genetic algorithms.
Experimental Protocol for Pharmaceutical Wastewater Treatment [30]:
- Adsorbent Preparation: A nano-filtration membrane is fabricated from palm sheath fiber, which is defatted and characterized using XRD to determine its crystalline composition.
- Batch Adsorption Experiments: A stock solution of Diclofenac Potassium is filtered through the membrane while varying four key process parameters: temperature (30–50 °C), pH (6–10), flow rate (1–5 ml/min), and initial concentration (40–120 mg/L).
- Model Development & Optimization:
- RSM: A statistical model is built to understand the influence and interactions of the four factors on removal efficiency.
- ANN: A network is trained on the experimental data to capture non-linear relationships.
- Genetic Algorithm: Used in conjunction with both models to find the parameter set that predicts the maximum removal efficiency.
- Validation: The optimized parameters are tested in triplicate experiments. The ANN model, which showed superior predictive accuracy, yielded an optimal removal efficiency of 84.78%, which was confirmed experimentally with an average efficiency of 84.67% [30].
Table 3: Comparison of RSM and ANN for Optimizing Diclofenac Potassium Removal [30]
| Metric | Response Surface Methodology (RSM) | Artificial Neural Network (ANN) |
|---|---|---|
| Correlation Coefficient (R²) | Strong correlation with data | Best predictive accuracy |
| Mean Absolute Error (MAE) | Higher than ANN | Lower than RSM |
| Absolute Average Relative Deviation (AARD) | Higher than ANN | Lower than RSM |
| Optimized Removal Efficiency | ~84% (inferred) | 84.78% (predicted), 84.67% (validated) |
| Key Advantage | Clear interpretation of factor interactions | Superior at capturing complex, non-linear relationships |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagent Solutions for Adsorption Studies
| Item Name | Function/Application | Specific Example |
|---|---|---|
| Quaternary Ammonium Functionalized AC | Electrostatic removal of PFAS from water | CTAB-impregnated Karanja shell carbon removed ~90-95% of short/long-chain PFCAs [31]. |
| Modified Clay Adsorbents | Low-cost removal of organic pollutants from wastewater | Basic activation & thermal treatment (750°C) of clay achieved 1199.93 mg/g capacity for Crystal Violet dye [32]. |
| Palm Sheath Fiber NF Membrane | Sustainable nano-filtration & adsorption | Used for pharmaceutical (Diclofenac) removal; characterized by XRD (75% calcite) [30]. |
| Al-Fumarate MOF | Advanced adsorbent for water capture/desalination | High water production capacity (23.5 m³/tonne/day) in adsorption desalination systems [33]. |
| Silver Nanoparticle SERS Substrates | Signal enhancement for trace contaminant detection | Enables detection of PFAS like PFOA down to femtogram per liter levels [3]. |
Visualizing Workflows and Signaling Pathways
Raman-DFT-ML Framework for Contaminant Detection
This diagram illustrates the integrated workflow for detecting environmental contaminants using Raman spectroscopy, DFT, and machine learning.
Multilevel Quantum Framework for Surface Chemistry
This diagram outlines the automated multilevel framework for achieving high-accuracy predictions of adsorption on ionic surfaces.
Leveraging DFT-Calculated Libraries for Contaminant Identification
The accurate identification of environmental contaminants is a cornerstone of public health and ecological safety. Traditional methods reliant on experimental reference spectra face significant challenges, including limited availability of chemical standards, spectral interference in complex matrices, and inability to keep pace with newly identified pollutants. Density Functional Theory (DFT)-calculated spectral libraries represent a transformative approach by providing in silico-generated reference data that can be systematically engineered to cover a vast chemical space. This guide objectively compares the performance of DFT-calculated libraries against traditional experimental libraries and other analytical approaches for contaminant identification, framing this comparison within the broader thesis that computational spectroscopy requires robust validation to achieve scientific acceptance.
The validation of DFT-calculated spectra sits at the intersection of computational chemistry, environmental science, and analytical technology. As regulatory frameworks struggle to keep pace with newly identified contaminants like polycyclic aromatic compounds (PACs) and per- and polyfluoroalkyl substances (PFAS), the ability to generate accurate theoretical spectra for compounds lacking commercial standards becomes increasingly vital. This comparison examines the experimental evidence supporting DFT's integration into mainstream environmental monitoring workflows.
Comparative Analysis: DFT-Calculated vs. Experimental Spectral Libraries
Performance Metrics Across Contaminant Classes
Table 1: Quantitative Performance Comparison of Identification Methods Across Contaminant Classes
| Contaminant Class | Identification Method | Key Performance Metrics | Limitations | Supporting Evidence |
|---|---|---|---|---|
| PFAS | DFT + Raman Spectroscopy | Strong similarity (>0.6) between DFT and experimental spectra; Differentiation of 9 PFAS by chain length/functional groups [3] | Requires validation for novel structures; Dependent on computational level | Experimental Raman spectra confirmed DFT predictions for 9 PFAS compounds; Unsupervised ML (PCA, t-SNE) enabled clear clustering [3] |
| PAHs/PACs | DFT + SERS + Machine Learning | High discriminative capability; Strong similarity values (>0.6) for multiple PAHs; Identification in complex soil matrices [5] | Challenging in low-concentration samples; Substrate-specific variations in SERS | Characteristic Peak Extraction (CaPE) algorithm isolated spectral features; CaPSim algorithm identified analytes robust to spectral shifts [5] |
| Protein Contaminants | Experimental Spectral Libraries | Increased protein identifications; Reduced false discoveries in DDA/DIA proteomics [34] | Limited to known contaminants; Requires physical samples | Implementation of contaminant FASTA and spectral libraries improved accuracy in bottom-up proteomics workflows [34] |
| Microbial Contaminants | Statistical Classification (decontam) | Effectively identified contaminant sequences in marker-gene and metagenomic data; Improved accuracy of microbial community profiles [35] | Primarily for external contaminants; Less effective for cross-contamination | Frequency-based and prevalence-based methods classified contaminants consistent with prior microscopic observations [35] |
Technical and Operational Characteristics
Table 2: Technical and Operational Comparison of Contaminant Identification Approaches
| Characteristic | DFT-Calculated Libraries | Traditional Experimental Libraries | Statistical Methods (e.g., decontam) |
|---|---|---|---|
| Coverage Scope | Virtually unlimited for structures that can be modeled; includes non-synthesized compounds [5] | Limited to commercially available or previously isolated compounds | Identifies study-specific contaminants based on patterns in experimental data [35] |
| Development Time | Rapid once computational framework established; dependent on computational resources | Time-consuming synthesis/purification; requires physical standards | Requires sequencing and control samples; analysis is rapid once data is collected |
| Cost Factors | High computational costs; minimal reagent/chemical costs | High costs for chemical standards, synthesis, and characterization | Moderate sequencing costs; minimal computational costs |
| Accuracy Limitations | Dependent on theoretical model accuracy; functional group performance varies | Gold standard when available; subject to experimental artifacts/impurities | Effective for external contaminants; limited for cross-contamination [35] |
| Implementation Complexity | Requires expertise in computational chemistry and spectral interpretation | Standardized protocols; accessible to most analytical laboratories | Accessible R package; integrates with existing bioinformatics workflows [35] |
| Environmental Application | Particularly valuable for persistent pollutants (PFAS, PAHs) and transformation products [5] [3] | Limited for emerging contaminants without available standards | Optimized for microbial community analysis in low-biomass environments [35] |
Experimental Protocols and Methodologies
DFT Spectral Calculation and Validation Workflow
The general methodology for developing and validating DFT-calculated spectral libraries follows a systematic workflow that integrates computational chemistry, experimental validation, and data analysis components.
Diagram 1: DFT Library Development Workflow
Protocol 1: DFT Spectral Calculation for Environmental Contaminants
This protocol outlines the key steps for generating DFT-calculated Raman spectra, as validated in PFAS and PAH detection studies [5] [3].
Molecular Structure Preparation
- Obtain initial molecular structures from databases like PubChem or create using molecular editing software
- For PFAS compounds: Systematic variation of chain length (C4-C12) and functional groups (carboxylic acid, sulfonic acid) as examined in recent studies [3]
Computational Parameters
- Employ Density Functional Theory with hybrid functionals (e.g., B3LYP) and basis sets (6-311G)
- Conduct geometry optimization followed by frequency calculations to ensure no imaginary frequencies
- Apply scaling factors (typically 0.96-0.98) to correct systematic overestimation of vibrational frequencies
Spectra Simulation
- Convert calculated frequencies to simulated spectra using Gaussian or Lorentzian broadening functions
- Incorporate instrumental parameters (resolution, laser wavelength) to match experimental conditions
Protocol 2: Experimental Validation of DFT-Calculated Spectra
Reference Standard Preparation
Spectral Acquisition
Data Processing and Comparison
- Apply preprocessing (background subtraction, normalization) to both experimental and calculated spectra
- Implement the Characteristic Peak Extraction (CaPE) algorithm to isolate distinctive spectral features [5]
- Calculate similarity metrics (CaPSim) between experimental and DFT-calculated spectra [5]
Machine Learning-Enhanced Identification Workflow
The integration of machine learning with DFT-calculated libraries creates a powerful framework for contaminant identification in complex environmental samples.
Diagram 2: ML-Enhanced Contaminant Identification
Protocol 3: Machine Learning Implementation for Contaminant Detection
Feature Extraction using Characteristic Peak Extraction (CaPE)
- Input: Raw SERS/Raman spectra from environmental samples
- Process: Identify prominent peaks while accommodating spectral shifts and amplitude variations common in SERS [5]
- Output: Characteristic spectral features for each sample
Pattern Recognition and Classification
Validation and Confidence Assessment
- Compare ML+DFT identifications with traditional methods (GC-MS) where available [5]
- Establish confidence metrics based on similarity scores and number of characteristic peaks matched
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for DFT-Validated Contaminant Detection
| Category | Specific Items | Function/Application | Example Use Cases |
|---|---|---|---|
| Computational Resources | DFT Software (Gaussian, ORCA), High-Performance Computing Cluster | Molecular modeling, geometry optimization, frequency calculations | Predicting Raman spectra for PFAS compounds with varying chain lengths [3] |
| Reference Materials | Certified PFAS/PAH Standards, Soil Samples, Solvents (HPLC grade) | Experimental validation of DFT predictions, method calibration | Creating controlled contamination samples for validation [5] |
| Spectral Enhancement | SERS Substrates (Au/Ag nanoparticles, nanoshells) | Signal amplification for trace-level detection | SiO₂ core-Au shell nanoparticles for PAH detection in soil extracts [5] |
| Instrumentation | Raman Spectrometer, GC-MS, FTIR | Spectral acquisition, reference analysis, method comparison | Experimental Raman measurements of 9 PFAS compounds [3] |
| Data Analysis Tools | Machine Learning Libraries (Python, R), Spectral Processing Software | Data preprocessing, feature extraction, pattern recognition | CaPE and CaPSim algorithms for spectral comparison [5] |
| Laboratory Consumables | Filters, Extraction Kits, Sample Preparation Materials | Environmental sample processing, contaminant extraction | Acetone extraction of PAHs from contaminated soil [5] |
The experimental data compiled in this comparison guide demonstrates that DFT-calculated libraries offer distinct advantages for identifying challenging environmental contaminants like PFAS and PAHs, particularly when commercial standards are unavailable. The validation framework establishing strong similarity (>0.6) between theoretical and experimental spectra provides a foundation for scientific acceptance of these computational approaches [5] [3].
While traditional experimental libraries remain the gold standard for established contaminants, DFT-calculated libraries excel in coverage of emerging contaminants and structural variants. The integration of machine learning with DFT predictions creates a powerful synergy that accommodates the real-world complexities of environmental samples. As computational resources continue to expand and theoretical methods refine, DFT-calculated libraries are positioned to become indispensable tools in the environmental analytical chemist's arsenal, ultimately accelerating the identification and monitoring of persistent environmental pollutants.
The detection and identification of polycyclic aromatic hydrocarbons (PAHs) in soil is a critical challenge in environmental science. These contaminants, known for their toxicity, persistence, and complex behavior in soil matrices, have traditionally required advanced laboratories and physical reference samples for accurate identification [36]. For many environmentally modified PAHs and their derivatives (PACs), which can be more toxic than their parent compounds, such reference standards are commercially unavailable or prohibitively expensive to synthesize [37] [38]. This case study examines a groundbreaking analytical framework that combines surface-enhanced Raman spectroscopy (SERS) with a virtual spectral library generated through density functional theory (DFT) and machine learning (ML) algorithms [36] [38]. We will objectively compare this in silico approach against conventional detection methods, presenting quantitative performance data and detailed experimental protocols to contextualize its performance within the broader validation of DFT-calculated spectra for environmental contaminant detection.
Background: The PAH Contamination Challenge
Polycyclic aromatic hydrocarbons are organic compounds containing multiple fused aromatic rings, produced primarily through incomplete combustion processes [39]. They are widely recognized for their toxic, mutagenic, and carcinogenic properties, posing significant risks to ecosystems and human health [40]. The U.S. Environmental Protection Agency has designated 16 PAHs as priority pollutants, though hundreds more exist in environmental samples, many lacking standardized detection methods [37] [39].
Soil acts as a primary sink for PAHs, where their detection is complicated by complex soil organic matter and the tendency of these compounds to undergo environmental transformations that alter their chemical structure and properties [36] [39]. Traditional remediation methods like thermal desorption, while effective, require precise efficiency predictions to avoid excessive energy use and costs [41], while nature-based solutions like phytoremediation demonstrate variable effectiveness across plant species [40].
Conventional PAH Detection Methods
Established Analytical Techniques
Traditional approaches for identifying PAHs in soil rely heavily on chromatographic separation coupled with various detection systems, primarily gas chromatography-mass spectrometry (GC-MS) or high-performance liquid chromatography (HPLC). These methods require advanced laboratory infrastructure, specialized personnel, and most significantly, physical reference standards for each target compound [36] [37]. The fundamental limitation of this approach lies in the lack of available standards for many PAH derivatives and transformation products that form under environmental conditions [36].
Experimental Limitations
The challenge extends beyond reference standard availability. As research on higher molecular weight PAHs has revealed, many compounds of significant toxicological concern, such as dibenzopyrene isomers, are not included in standard monitoring protocols due to the prohibitive cost and complexity of their synthesis and purification [37]. Furthermore, environmental samples frequently contain emission peaks that don't correspond to any commercially available standards, creating significant gaps in contamination assessment [37].
The In Silico Spectra Approach: Methodology and Workflow
Core Technological Framework
The innovative approach developed by researchers at Rice University and Baylor College of Medicine integrates three complementary technologies to overcome traditional detection limitations [36] [38]:
Surface-Enhanced Raman Spectroscopy (SERS): A light-based imaging technique that analyzes how light interacts with molecules, generating unique spectral "fingerprints" for each compound. The method uses specially designed signature nanoshells to enhance relevant traits in the spectra obtained from soil samples [36].
Density Functional Theory (DFT) Calculations: A computational modeling approach that predicts the molecular structure and electronic properties of PAHs and PACs, enabling the generation of theoretical Raman spectra without needing physical samples [36] [38]. This creates a virtual spectral library of "chemical fingerprints" for compounds that have never been isolated or studied experimentally [36].
Machine Learning Algorithms: A two-stage physics-informed ML pipeline consisting of:
- Characteristic Peak Extraction (CaPE): Isolates distinctive spectral features from complex sample data [36] [38].
- Characteristic Peak Similarity (CaPSim): Identifies analytes with high robustness to spectral shifts and amplitude variations, matching experimental observations with theoretical predictions [36] [38].
Experimental Protocol
The methodology was rigorously validated through controlled experiments [36]:
- Soil Preparation: Researchers tested the method on soil from a restored watershed and natural area, using both artificially contaminated samples and uncontaminated controls.
- SERS Analysis: Soil samples were analyzed using surface-enhanced Raman spectroscopy with customized nanoshells to enhance spectral features.
- Spectral Matching: The machine learning pipeline parsed relevant spectral traits from real-world soil samples and matched them to compounds in the virtual DFT-calculated library.
- Validation: Similarity values exceeding 0.6 were established as a threshold for positive identification, confirming strong correlation between DFT-calculated and experimental SERS spectra for multiple PAHs [38].
The diagram below illustrates the integrated workflow of this in silico detection approach:
Research Reagent Solutions
The table below details essential materials and computational tools required for implementing this in silico detection methodology:
Table 1: Research Reagent Solutions for In Silico PAH Detection
| Component Category | Specific Tools/Materials | Function in Workflow |
|---|---|---|
| Spectroscopic Equipment | Portable Raman Spectrometer with SERS Nanoshells | Enhances spectral signals from soil samples for analysis [36] |
| Computational Software | DFT Modeling Packages (e.g., Gaussian) | Predicts molecular structures and calculates theoretical spectra [37] |
| Machine Learning Algorithms | Characteristic Peak Extraction (CaPE) & Characteristic Peak Similarity (CaPSim) | Isolates and matches spectral features to virtual library [36] [38] |
| Spectral Library | DFT-Calculated PAH Spectral Database | Provides reference "fingerprints" for identification without physical standards [36] |
| Soil Processing Tools | Standardized Soil Sampling and Preparation Kits | Ensures consistent sample quality for reliable spectroscopic analysis [36] |
Performance Comparison: In Silico vs. Conventional Methods
Quantitative Performance Metrics
The table below presents a structured comparison of key performance indicators between conventional detection methods and the in silico spectra approach:
Table 2: Performance Comparison of PAH Detection Methods
| Performance Parameter | Conventional Methods | In Silico Spectra Approach |
|---|---|---|
| Reference Dependency | Requires physical reference samples [36] | Uses DFT-calculated virtual libraries [36] [38] |
| Detection Capability | Limited to commercially available standards [37] | Identifies unisolated/modified compounds [36] |
| Spectral Similarity Score | N/A (physical standards) | >0.6 for validated PAHs [38] |
| Implementation Flexibility | Laboratory-dependent [36] | Potential for portable field deployment [36] |
| Theoretical Foundation | Empirical measurements only [36] | Integrates theoretical physics with experimental data [36] [38] |
| Environmental Relevance | Limited to parent compounds [36] | Detects transformed derivatives [36] |
Detection Accuracy and Validation
The in silico method demonstrated reliable identification of even minute traces of PAHs in contaminated soil samples, with the machine learning pipeline successfully matching experimental spectra to DFT-calculated references [36]. Researchers reported "strong similarity values (>0.6)" between DFT-calculated and experimental Surface-Enhanced Raman Spectra for multiple PAHs, confirming the accuracy and discriminative capability of the approach [38]. This performance is particularly notable given that the method successfully identified compounds without experimental reference data, including "those formed through environmental modification of PAHs" [38].
Advantages and Limitations in Research Applications
Technological Advantages
The in silico spectra approach offers several distinct advantages for environmental research and monitoring:
- Comprehensive Contaminant Screening: By moving beyond dependency on physical standards, the method enables detection of previously unidentifiable PAH derivatives and transformation products that form in soil environments [36] [38].
- Theoretical Prediction Capability: As theorized by Senftle, "on the theory side, we can predict what the picture will look like" [36], enabling the method to account for environmental transformations of PAHs over time.
- Field Deployment Potential: The integration of machine learning algorithms and theoretical spectral libraries with portable Raman devices creates a pathway toward mobile detection systems that could provide rapid on-site analysis without laboratory delays [36].
Current Limitations and Research Directions
While promising, the methodology has limitations that require further research and development:
- Computational Demands: DFT calculations for complex molecules require significant computational resources, potentially limiting accessibility for some research groups [37].
- Validation Scope: While successfully validated for multiple PAHs, the approach requires further testing across a broader range of soil types and contamination scenarios to establish universal applicability [36].
- Spectral Interpretation Complexity: The machine learning pipeline, while robust, requires specialized expertise to implement and optimize for novel contaminant classes [36] [38].
Implications for Environmental Monitoring and Research
This in silico detection framework represents a paradigm shift in environmental contaminant analysis. By combining first-principles physics calculations with advanced machine learning and spectroscopic techniques, it addresses a critical gap in environmental monitoring capabilities [36] [38]. The approach is particularly valuable for identifying toxic PAH derivatives that have evaded traditional detection methods due to the lack of reference standards.
The methodology also shows significant promise for predictive environmental assessment. As demonstrated in parallel research on thermal desorption efficiency prediction using machine learning [41], computational approaches are increasingly capable of modeling complex environmental processes. The in silico spectra approach extends this capability to the fundamental identification stage, potentially enabling more comprehensive risk assessment and remediation planning for contaminated sites.
Future developments in this field will likely focus on expanding virtual spectral libraries, optimizing machine learning algorithms for greater discrimination between structurally similar compounds, and integrating the approach with complementary detection methodologies for validation. As computational power increases and spectroscopic technologies become more portable, this integrated approach may eventually become standard practice for environmental monitoring and regulatory compliance.
The case study demonstrates that the in silico spectra approach for detecting PAHs in soil represents a significant advancement over conventional methods. By leveraging density functional theory to create virtual spectral libraries and machine learning to match experimental observations, this methodology overcomes the fundamental limitation of reference standard dependency that has constrained environmental monitoring. Validation results confirm its ability to reliably identify both known PAHs and previously undetectable transformation products, with similarity values exceeding 0.6 for multiple compounds [38].
While conventional chromatographic methods remain essential for quantitative analysis, the in silico approach offers unparalleled capabilities for comprehensive contaminant screening and identification. Its development marks important progress in validating computational spectroscopy for practical environmental applications, providing researchers and environmental professionals with a powerful new tool for assessing and addressing soil contamination by polycyclic aromatic hydrocarbons and their derivatives.
Navigating Challenges: Overcoming DFT Limitations in Environmental Systems
Accurately modeling intermolecular interactions, particularly dispersion forces and charge transfer, represents a fundamental challenge in computational chemistry with significant implications for applied environmental science. The validation of Density Functional Theory (DFT)-calculated spectra hinges on properly accounting for these complex electronic interactions. Failure to accurately describe the interplay between long-range dispersion and charge transfer can lead to substantial errors in predicting molecular adsorption geometries, energy level alignment, and ultimately, the interpretation of spectroscopic data used for contaminant identification. This guide provides a comparative analysis of computational and experimental approaches, highlighting common failure modes and solutions for researchers working at the intersection of computational chemistry and environmental contaminant detection.
Theoretical Foundations and Computational Failure Modes
The Interplay of Dispersion and Charge Transfer
Dispersion forces and charge transfer interactions collectively govern the behavior of molecules at interfaces, yet they present distinct challenges for computational modeling. Dispersion interactions are weak, attractive forces arising from correlated electron density fluctuations between molecules, while charge transfer involves the actual movement of electron density between chemical species. The strong interplay between these phenomena is particularly pronounced at metal-organic interfaces, where both effects significantly stabilize the system [42].
When molecules adsorb onto metal surfaces, the exchange of charge modifies their electronic properties and atomic polarizabilities. This creates a complex feedback loop: charge transfer alters polarizability, which in turn affects dispersion interactions. Standard computational methods often treat these effects independently, leading to inaccurate predictions of key properties like adsorption heights and binding energies [42].
Common DFT Failure Modes
Density Functional Theory, while widely used, exhibits several systematic failures in handling dispersion and charge transfer:
Inadequate Adsorption Geometry Prediction: Recent studies demonstrate that dispersion-inclusive DFT methods fail to correctly capture adsorption heights for strong donors like alkali atoms on silver surfaces, with errors exceeding experimental uncertainty [42].
Polarizability Miscalibration: The core issue stems from the inability of standard methods to account for changes in atomic polarizability due to charge transfer. The fixed dispersion parameters in most DFT functionals cannot adapt to the modified electronic environment of charged systems [42].
Compensating Error Propagation: The tendency of errors in dispersion and charge transfer calculations to offset each other creates false positives in method validation, where apparently correct energies mask incorrect physical descriptions.
Table 1: Common DFT Failure Modes in Dispersion and Charge Transfer Modeling
| Failure Mode | Physical Origin | Impact on Predictions | Systematic Error |
|---|---|---|---|
| Incorrect adsorption heights | Fixed dispersion parameters unresponsive to charge transfer | Errors in interfacial structure (>0.1 Å) | Underestimation of bonding distances |
| Band alignment errors | Improper charge redistribution at interface | Incorrect energy level alignment (>0.2 eV) | Overestimation of charge injection barriers |
| Polarizability miscalibration | Neglect of electron density modification | Faulty dispersion energy scaling (>15%) | Underbinding for donors, overbinding for acceptors |
Comparative Analysis of Computational Approaches
Dispersion-Corrected DFT Methods
The development of dispersion-inclusive DFT approaches has significantly improved the description of weak interactions, yet significant challenges remain:
Van der Waals Functionals: Methods such as the vdW-DF family incorporate non-local correlation to capture dispersion. While generally improving binding energy predictions, they still struggle with charge-transfer systems where polarizability changes occur.
Empirical Dispersion Corrections: Grimme's DFT-D methods add an empirical R⁻⁶ term to account for dispersion. These approaches are computationally efficient but rely on fixed parameters that don't adapt to charge-induced polarizability changes [42].
Self-Consistent Polarizability Scaling: Emerging approaches address fundamental limitations by rescaling dispersion parameters based on calculated atomic charges, directly addressing the polarizability-change failure mode [42]. This method has demonstrated improved accuracy for alkali-organic metal-organic frameworks on silver surfaces.
Beyond Standard DFT: COSMO-RS and Specialized Methods
Recent advances in thermodynamic property prediction have led to improved handling of dispersion interactions:
openCOSMO-RS Enhancements: The implementation of a new dispersion term based on atomic polarizabilities in openCOSMO-RS represents a significant improvement over previous parameterizations. This approach reduces the number of adjustable parameters while increasing accuracy across diverse mixture types [43].
Atomic Polarizability Descriptors: Using atomic polarizabilities as fundamental descriptors for dispersion interactions has shown promise for predictive thermodynamic models, particularly for halocarbon systems and complex mixtures relevant to environmental sampling [43].
Table 2: Performance Comparison of Computational Methods for Dispersion/Charge Transfer Systems
| Method | Dispersion Treatment | Charge Transfer Adaptability | Accuracy for Adsorption Heights | Computational Cost |
|---|---|---|---|---|
| Standard DFT (GGA) | None | None | Poor (>0.3 Å error) | Low |
| DFT-D2/D3 | Empirical correction | Limited (fixed parameters) | Moderate (0.1-0.2 Å error) | Low |
| vdW-DF | Non-local functional | Moderate (via electron density) | Moderate (0.1-0.2 Å error) | Medium |
| Rescaled Dispersion | Scaled empirical | High (polarizability rescaling) | Good (<0.1 Å error) [42] | Low-Medium |
| openCOSMO-RS (new) | Atomic polarizability-based | Moderate (via segment charges) | N/A (for thermodynamics) | Low |
Experimental Validation Techniques
Electron Ptychography for Charge Density Measurement
Validating computational predictions of charge transfer requires direct experimental measurement of electron density changes with exceptional sensitivity:
Principle of Operation: Electron ptychography uses a focused electron beam scanned across a sample with overlapping illumination positions. The resulting diffraction patterns are processed via phase retrieval algorithms to reconstruct the electron density and potential with sub-Ångstrom resolution [44] [45].
Detection of Charge Transfer: In monolayer WS₂, ptychography has directly imaged charge transfer from tungsten to sulfur sites, revealing a ~10% difference in charge density compared to the independent atom model [44] [45]. This provides quantitative validation for DFT predictions of bonding-induced charge redistribution.
Advantages over Conventional STEM: Unlike annular dark-field imaging, which is dominated by nuclear scattering, ptychographic phase imaging is directly sensitive to the electric potential, enabling charge transfer visualization [45]. The method's inherent dose efficiency also makes it suitable for radiation-sensitive materials.
Spectroscopic Validation of DFT-Calculated Spectra
DFT-calculated infrared absorption spectra provide critical templates for identifying environmental contaminants, but require careful validation:
Protocol for Spectral Prediction: DFT calculations using software like Gaussian can predict IR spectra for target molecules, such as nitrosamines in water, by computing vibrationally excited states within a continuous solvation model [46].
Experimental Correlation: Calculated spectra must be correlated with laboratory measurements to establish reliability. For nitrosamines, this approach has provided proof-of-concept for practical detection in environmental samples [46].
Limitations and Considerations: The accuracy of DFT-calculated spectra depends heavily on the functional selection, basis set completeness, and solvation model appropriateness. Systematic errors often arise from anharmonic effects not captured by standard calculations.
Applications in Environmental Contaminant Detection
Machine Learning-Enhanced Contaminant Identification
Innovative approaches combining DFT calculations with machine learning have recently emerged for detecting environmental pollutants:
Virtual Spectral Libraries: DFT calculations generate theoretical spectra for pollutants that may lack experimental reference data, creating "virtual fingerprints" for compounds like polycyclic aromatic hydrocarbons (PAHs) and their derivatives [36].
Machine Learning Matching: Characteristic peak extraction and similarity algorithms parse relevant spectral traits from real-world samples and match them to the computationally generated library, enabling identification of chemicals without experimental reference standards [36].
Field Deployment Potential: This DFT-ML framework can be integrated with portable Raman devices, potentially enabling on-site detection of hazardous compounds without laboratory analysis [36].
Comprehensive Analytical Protocols for Complex Mixtures
Advanced analytical methods for environmental monitoring must address the challenge of detecting diverse contaminants with varying physicochemical properties:
Multi-Residue Extraction Methods: Novel protocols now enable quantification of 285 organic air pollutants spanning polar and non-polar compound classes, including amines, organic acids, pesticides, phenols, PAHs, and PCBs [47].
Sample Preparation Optimization: Accelerated solvent extraction (ASE) combined with solid-phase extraction (SPE) provides efficient recovery of diverse analytes. Derivatization with reagents like MtBSTFA enhances volatility and stability for GC-MS analysis [47].
Adsorbent Material Advances: Nitrogen-doped carbon-coated silicon carbide foam (NMC@SiC) passive samplers offer improved surface area and tunable chemistry for capturing both polar and non-polar compounds compared to traditional polyurethane foam [47].
Table 3: Research Reagent Solutions for Dispersion and Charge Transfer Studies
| Reagent/Platform | Function | Application Context | Key Advantage |
|---|---|---|---|
| Nitrogen-doped carbon-coated SiC foam (NMC@SiC) | Passive air sampler adsorbent | Broad-spectrum pollutant capture [47] | Enhanced surface area, tunable chemistry for polar/non-polar compounds |
| MtBSTFA derivatization reagent | Silylation of polar functional groups | GC-MS analysis of amines, acids, phenols [47] | Improves volatility, stability, and detection sensitivity |
| Nano-energetic materials (nEMs) | Controlled pressure pulse generation | Shock-induced dispersion studies [48] | Laboratory-scale simulation of explosive dispersion patterns |
| Viton B binder | Reactive composite fabrication | nEM preparation for dispersion experiments [48] | Stable binder for fuel-oxidizer composites |
| Hydrophobic silica (K-T30) | Powder coating for cohesion control | Powder flowability modification [48] | Tunable interparticle cohesion while maintaining other properties |
Integrated Workflow for Method Validation
The synergy between computational prediction and experimental validation enables robust detection of environmental contaminants. The following workflow integrates the approaches discussed in this guide:
The accurate description of dispersion forces and charge transfer remains challenging for computational methods, with common failure modes including incorrect adsorption geometries and miscalibrated polarizability effects. Rescaling dispersion parameters based on charge transfer and incorporating atomic polarizabilities represent promising approaches to address these limitations. Experimental techniques like electron ptychography provide crucial validation by directly imaging charge redistribution at the atomic scale. For environmental detection applications, integrating DFT-calculated spectra with machine learning enables identification of contaminants without experimental reference standards. As computational methods continue to improve their treatment of these complex interactions, and experimental validation techniques become more sensitive, the reliability of predictive models for environmental contaminant behavior will correspondingly advance, enabling more effective monitoring and remediation strategies.
Accurate detection and characterization of environmental contaminants, such as per- and polyfluoroalkyl substances (PFAS), represent a significant challenge in environmental chemistry. These persistent pollutants, with their strong carbon-fluorine bonds and complex molecular structures, necessitate advanced analytical techniques for precise identification and monitoring [3]. Among these, vibrational spectroscopic methods like Raman spectroscopy have emerged as powerful tools, particularly when complemented by computational predictions from Density Functional Theory (DFT). The reliability of these computational predictions, however, hinges critically on the appropriate selection of two fundamental components: the exchange-correlation functional and the basis set. This guide provides a systematic benchmarking approach for these selections, specifically framed within the validation of DFT-calculated spectra for environmental contaminant detection research. We present objective comparisons of performance and supporting experimental data to empower researchers in making informed computational choices that balance accuracy with efficiency.
Theoretical Framework: Understanding Basis Sets and Functionals
Density Functional Theory Fundamentals
Density Functional Theory provides the theoretical foundation for calculating molecular structures, energies, and properties by determining the electron density rather than dealing with the many-electron wavefunction. In the Kohn-Sham formulation, the energy is expressed as:
E~KS~ = V + 〈hP〉 + 1/2〈PJ(P)〉 + E~X~[P] + E~C~[P]
where V represents nuclear repulsion, 〈hP〉 the one-electron energy, 1/2〈PJ(P)〉 the classical Coulomb repulsion, and E~X~[P] and E~C~[P] the exchange and correlation functionals, respectively [49]. The accuracy of a DFT calculation depends critically on the mathematical expressions chosen for E~X~[P] and E~C~[P] (the "functional") and the set of basis functions used to expand the Kohn-Sham orbitals (the "basis set").
Basis Sets in Quantum Chemistry
A basis set is a collection of mathematical functions (basis functions) centered on atoms, used to represent the molecular orbitals. In Gaussian-type orbital approaches, these are typically contracted Gaussian-type functions [50]. The most basic classification of basis sets includes:
- Minimal Basis Sets (e.g., STO-3G): Use the minimum number of functions needed for each atom, suitable for preliminary calculations [51].
- Split-Valence Basis Sets (e.g., 3-21G, 6-31G): Use different basis functions for core and valence electrons, providing improved accuracy over minimal basis sets [51].
- Polarized Basis Sets (e.g., 6-31G(d), 6-31G): Add functions with higher angular momentum (d, f orbitals) to better describe electron distribution distortions [51].
- Diffuse Functions (e.g., 6-31+G, 6-31++G): Include functions with small exponents to better describe electrons far from the nucleus, important for anions and weak interactions [51].
- Correlation-Consistent Basis Sets (e.g., cc-pVDZ, cc-pVTZ, cc-pVQZ): Systematically constructed to converge toward the complete basis set limit, with the nomenclature indicating double, triple, and quadruple-zeta quality [51].
Benchmarking Methodologies: Protocols for Validation
General Workflow for Computational Benchmarking
The validation of computational methods requires systematic comparison against reliable experimental data or high-level theoretical references. The following diagram illustrates a robust workflow for benchmarking basis sets and functionals specifically for spectroscopic applications in environmental contaminant research.
Case Study: PFAS Spectroscopic Characterization
Recent research on PFAS compounds provides an exemplary model for benchmarking protocols. In one comprehensive study, researchers measured experimental Raman spectra of nine PFAS compounds with varying chain lengths and functional groups, including perfluoroheptanoic acid (PFHpA), perfluorooctanoic acid (PFOA), and perfluorodecanoic acid (PFDA) [3]. These compounds were selected to represent structures relevant to environmental contamination, as listed in the U.S. Environmental Protection Agency's Draft Method 1633.
The computational methodology employed density functional theory calculations with various functionals and basis sets to predict vibrational frequencies and intensities. The specific workflow included:
- Molecular Structure Preparation: Molecular structures of PFAS compounds were built and initially optimized [3].
- Geometry Optimization: Full geometry optimization was performed using selected density functionals with polarized basis sets [3].
- Frequency Calculations: Vibrational frequency calculations were performed on optimized structures to obtain theoretical Raman spectra [3].
- Spectral Comparison: Theoretical spectra were compared against experimental Raman measurements through frequency matching and intensity pattern analysis [3].
- Statistical Validation: Quantitative metrics including mean absolute deviation (MAD) between experimental and calculated frequencies were computed [3].
- Multivariate Analysis: Unsupervised machine learning techniques, specifically Principal Component Analysis (PCA) and t-distributed Stochastic Neighbor Embedding (t-SNE), were applied to cluster and classify PFAS compounds based on their spectral features [3].
Case Study: Flavins and Resonance Raman Spectroscopy
A separate extensive benchmark study focused on resonance Raman spectroscopy of lumiflavin, a model system for flavin cofactors, providing robust protocols for functional selection under resonance conditions [9]. This study evaluated 42 DFT functionals against experimental Evolution Associated Spectra (EAS) of FMN, considering multiple validation criteria:
- Excitation Energy Accuracy: Percent error of calculated 0-0 transition energies compared to experimental values [9].
- Spectral Correlation: Percent error of correlation between experimental and calculated resonance Raman spectra [9].
- Resonance Enhancement Impact: Difference in percent errors between off-resonance and resonance Raman correlations [9].
- Intensity Reproduction: Accuracy in predicting resonance Raman intensity of key experimental peaks [9].
- Visual Spectral Agreement: Qualitative assessment of whether theoretical spectral profiles matched experimental patterns [9].
This comprehensive approach employed the cc-pVDZ basis set and its augmented version (aug-cc-pVDZ) throughout, allowing focus on functional performance while maintaining applicability to larger systems like protein environments [9].
Performance Comparison of Density Functionals
Quantitative Benchmarking Data
The table below summarizes performance data for selected density functionals from recent benchmarking studies, highlighting their accuracy for spectroscopic predictions relevant to environmental contaminant research.
Table 1: Performance Benchmarking of Density Functionals for Vibrational Spectroscopy
| Functional | Type | Key Features | Test System | Performance Metrics |
|---|---|---|---|---|
| B3LYP [49] [9] | Hybrid GGA | 20% HF exchange; widely used | Flavins, PFAS | Good excitation energies; moderate vibrational accuracy [9] |
| HCTH [9] | Pure GGA | No HF exchange | Flavins | Top performer for resonance Raman; accurate frequencies [9] |
| τ-HCTH [52] | Meta-GGA | Includes kinetic energy density | Isotopic Fractionation | MAD: 22‰ (D/H), 4.1‰ (heavy atoms) [52] |
| OLYP [9] | Pure GGA | Handy-Cohen correlation | Flavins | Excellent resonance Raman correlation [9] |
| TPSSh [9] | Hybrid Meta-GGA | 10% HF exchange | Flavins | Strong resonance Raman performance [9] |
| O3LYP [52] | Hybrid GGA | Optimized exchange weighting | Isotopic Fractionation | MAD: 21‰ (D/H), 3.9‰ (heavy atoms) [52] |
| wB97XD [49] | Long-range corrected | Includes dispersion; range-separated | General Purpose | Good for excited states & weak interactions [49] |
| CAM-B3LYP [49] | Long-range corrected | Attenuated exchange; range-separated | General Purpose | Improved charge transfer excitations [49] |
| LC-ωPBE [49] | Long-range corrected | Full range separation | General Purpose | Accurate for high orbitals & excitations [49] |
| PBE1PBE (PBE0) [49] | Hybrid GGA | 25% HF exchange | General Purpose | Good all-purpose hybrid functional [49] |
Interpretation of Functional Performance
The benchmarking data reveals several important patterns for functional selection:
GGA Functionals for Vibrational Frequencies: Pure generalized gradient approximation (GGA) functionals like HCTH and OLYP demonstrated exceptional performance for resonance Raman spectroscopy of flavin systems, outperforming many more complex hybrid functionals [9]. This suggests that for ground-state vibrational frequencies and resonance Raman applications, sophisticated treatments of exchange may be less critical than proper description of correlation.
Hybrid Functionals for Mixed Properties: Hybrid functionals like B3LYP remain popular choices for general-purpose computational studies, particularly when balancing accuracy for multiple properties including structures, energies, and spectroscopic predictions [9].
Specialized Functionals for Specific Applications: The strong performance of O3LYP for calculating equilibrium isotopic fractionation, with mean absolute deviations of 21‰ for D/H fractionation and 3.9‰ for heavy-atom fractionation, highlights how certain functionals may be particularly well-suited for specific applications in environmental research [52].
Long-Range Corrections for Excited States: For properties involving electronic excitations, such as those relevant to resonance Raman spectroscopy, long-range corrected functionals like LC-ωPBE and CAM-B3LYP provide improved performance for charge-transfer transitions and high-lying orbitals [49].
Performance Comparison of Basis Sets
Quantitative Basis Set Benchmarking
The table below presents performance and computational cost data for commonly used basis sets, particularly relevant for spectroscopic studies of environmental contaminants.
Table 2: Performance and Computational Cost of Selected Basis Sets
| Basis Set | Type | Total Cartesian Functions (Tryptophan) | Relative CPU Time (B3LYP) | Key Applications & Notes |
|---|---|---|---|---|
| 6-31G [53] | Split-Valence Double-Zeta | 159 | 1.0x (Reference) | Initial optimizations; small systems |
| 6-31+G [53] | Diffuse Augmented DZ | 219 | 3.3x | Anions, weak interactions; recommended for frequency calculations [53] |
| 6-31+G(d,p) [53] | Polarized & Diffuse DZ | 345 | 7.7x | General purpose spectroscopy; good accuracy/cost balance [53] |
| cc-pVDZ [51] [53] | Correlation-Consistent DZ | 285 | 3.8x | High-quality double-zeta; systematically improvable [51] |
| cc-pVTZ [51] | Correlation-Consistent TZ | - | ~10-20x (Est.) | High-accuracy applications; reference calculations |
| aug-cc-pVDZ [51] [9] | Augmented cc-pVDZ | - | ~5x (Est.) | Improved excited states & anion description [9] |
| def2-TZVP [52] | Triple-Zeta Valence Polarized | - | ~5-10x (Est.) | Excellent for isotopic fractionation with O3LYP [52] |
| LanL2DZ [51] | Effective Core Potential | - | Varies | Heavy elements; replaces core electrons with potentials [51] |
Interpretation of Basis Set Performance
The benchmarking data reveals several important considerations for basis set selection:
Balancing Cost and Accuracy: For the tryptophan molecule, moving from 6-31G to 6-31+G(d,p) increased basis function count from 159 to 345, with computational time increasing approximately 7.7-fold for B3LYP calculations [53]. This highlights the importance of selecting basis sets that provide sufficient accuracy while remaining computationally feasible, especially for larger systems like environmental contaminants.
Polarization and Diffuse Functions: The addition of polarization functions (d, p) is crucial for properly describing molecular deformations, while diffuse functions (+) are important for modeling weak interactions, anions, and excited states - all potentially relevant for environmental contaminant behavior [51].
Systematically Improvable Basis Sets: Correlation-consistent basis sets (cc-pVXZ) offer a systematic path to the complete basis set limit through increasing levels of X (D, T, Q, 5, 6), making them valuable for high-accuracy reference calculations [51].
Adequate but Affordable Basis Sets: For many applications, particularly with larger molecules, polarized double-zeta basis sets like 6-31+G(d,p) or cc-pVDZ provide the best balance of accuracy and computational efficiency [53] [9].
Table 3: Essential Computational Tools for Spectroscopic Benchmarking
| Tool/Resource | Function | Application Notes |
|---|---|---|
| Gaussian 16 [51] [49] | Quantum Chemistry Package | Implements wide range of DFT methods, basis sets, spectroscopic properties [51] |
| def2-TZVP [52] | Triple-Zeta Basis Set | Shows excellent performance for isotopic fractionation with O3LYP functional [52] |
| Polarizable Continuum Model (PCM) [9] | Solvation Method | Models solvent effects; crucial for environmental applications [9] |
| UltraFine Integration Grid [49] | DFT Numerical Grid | Default in Gaussian 16; enhances calculation accuracy [49] |
| FREQ Program [9] | Frequency Scaling | Generates frequency scaling factors for improved agreement with experiment [9] |
| Principal Component Analysis (PCA) [3] | Multivariate Analysis | Clusters and classifies spectral data; identifies patterns [3] |
| t-Distributed Stochastic Neighbor Embedding (t-SNE) [3] | Dimensionality Reduction | Visualizes high-dimensional spectral data; reveals clustering [3] |
Integrated Workflow: From Calculation to Environmental Application
The relationship between computational choices and their impact on predicting environmentally relevant properties is summarized in the following workflow, which integrates basis set and functional selection with specific environmental applications.
Based on the comprehensive benchmarking data presented, we can derive specific recommendations for computational method selection in environmental contaminant research:
For vibrational spectroscopy applications including Raman characterization of PFAS and similar contaminants, the HCTH, OLYP, and TPSSh functionals provide excellent accuracy based on rigorous benchmarking against experimental data [9]. When paired with the def2-TZVP basis set, these functionals offer an optimal balance of computational cost and predictive accuracy for environmental applications.
For isotopic fractionation studies, particularly relevant for tracking contaminant transformation and degradation pathways, the O3LYP functional with def2-TZVP basis set demonstrated the lowest mean absolute deviations in benchmark studies [52].
For general-purpose spectroscopic characterization of environmental contaminants, hybrid functionals like B3LYP and PBE0 with polarized double-zeta basis sets such as 6-31+G(d,p) or cc-pVDZ provide reliable performance with reasonable computational cost [53] [9].
The integration of computational predictions with experimental validation, supplemented by multivariate analysis techniques like PCA and t-SNE, creates a powerful framework for advancing environmental detection and monitoring capabilities [3]. By following the systematic benchmarking approaches outlined in this guide, researchers can make informed decisions about computational methods that generate reliable, predictive results for addressing challenging environmental contamination problems.
Density Functional Theory (DFT) serves as a cornerstone in computational chemistry, enabling the prediction of molecular structures, reaction energies, and spectroscopic properties. However, conventional density functional approximations (DFAs) contain intrinsic systematic errors that limit their predictive accuracy for complex chemical systems. In the critical field of environmental contaminant detection, where computational methods guide the identification of pollutants like pesticides and per- and polyfluoroalkyl substances (PFAS), these errors can significantly impact reliability. This guide compares the leading approaches for correcting systematic DFT errors, with a specific focus on validating DFT-calculated spectra for environmental applications.
Understanding Systematic Errors in DFT
Despite the formal exactness of DFT, practical calculations employ DFAs that suffer from delocalization error and improper description of dispersion (van der Waals) interactions [54]. These systematic errors manifest as significant inaccuracies in computed formation enthalpies—often several hundred meV/atom for compounds involving transition metals or localized electronic states [55]. For spectroscopic applications, these errors can alter predicted vibrational frequencies and peak intensities, potentially leading to misidentification of environmental contaminants.
The recognition that semi-local density functionals do not properly capture dispersion interactions represented a major development in DFT during the mid-2000s [56]. Simultaneously, delocalization error remains a key challenge that conventional DFAs fail to address for critical physical properties [54]. These limitations necessitate correction protocols to achieve the accuracy required for reliable environmental detection methodologies.
Comparison of Correction Approaches
Two principal philosophies have emerged for addressing DFT's systematic errors: empirical dispersion corrections and scaling correction methods. The table below summarizes their key characteristics, performance, and ideal use cases.
Table 1: Comparison of Major DFT Correction Methods
| Method Category | Specific Methods | Key Features | Accuracy Improvement | Best For |
|---|---|---|---|---|
| Empirical Dispersion Corrections | DFT-D2, DFT-D3, DFT-D4 [56] | - Adds empirical potentials (e.g., -C₆/R⁶)- Parameterized for specific elements- Multiple versions with different damping functions | Reduces errors in formation enthalpies to ~50 meV/atom or less [55] | General thermochemistry, non-covalent interactions, organometallic systems |
| Scaling Corrections | Global Scaling Correction (GSC), Localized Orbital Scaling Correction (LOSC) [54] | - Targets delocalization error systematically- Improves orbital energies and quasiparticle spectra- Enables better prediction of excited states | Accurately predicts quasiparticle energies and photoemission spectra [54] | Excited-state problems, charge transfer excitations, polymer polarizability |
Experimental Protocols for Method Validation
Workflow for Spectroscopic Validation of PFAS Compounds
Research combining Raman spectroscopy with machine learning for PFAS detection establishes a robust protocol for validating DFT methodologies [28]. The workflow proceeds through these critical stages:
- Sample Preparation and Spectral Acquisition: Nine PFAS compounds with varying functional groups and chain lengths are selected. Raman spectra are collected across low, medium, high, and ultra-high wavenumber regions to capture distinct vibrational peaks.
- DFT Calculations with Appropriate Corrections: Density functional theory calculations model the electronic structure of each PFAS compound. Dispersion corrections are essential to properly account for intermolecular interactions.
- Vibrational Mode Assignment: Theoretical spectra from DFT are used to associate experimental vibrational peaks with specific molecular motions, confirming the physical basis for spectral signatures.
- Machine Learning Classification: Advanced data analysis techniques, including principal component analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE), classify and separate Raman spectra based on structural features.
- Similarity Assessment: Processed experimental spectra are quantitatively compared to DFT-calculated reference spectra to validate the accuracy of the computational methodology.
Protocol for Pesticide Identification Using DFT-D
A separate study establishing a theoretical Raman database for 166 pesticides provides another exemplary validation protocol [57]:
- Database Construction: DFT calculations, including dispersion corrections, generate theoretical Raman spectra for 166 pesticides, focusing on analyzing Raman peaks and vibrational modes.
- Isomer Analysis: The effects of functional group isomers and chain isomers on spectral features are systematically explored to understand structural impacts on spectra.
- Unsupervised Machine Learning: PCA and t-SNE algorithms are applied to identify the 22 heterocyclic pesticides without prior labeling.
- Sensitivity Enhancement: Surface-Enhanced Raman Spectroscopy (SERS) substrates are employed to significantly enhance detection sensitivity for practical application.
Diagram 1: DFT Spectral Validation Workflow for Environmental Contaminants
Quantitative Performance Assessment
The performance of various DFT methodologies can be quantitatively assessed through their impact on formation enthalpy accuracy and spectroscopic prediction. The table below summarizes key performance metrics from benchmark studies.
Table 2: Quantitative Performance of DFT Correction Methods
| Functional/Correction | Basis Set | Mean Absolute Error (Formation Enthalpy) | Spectral Prediction Accuracy | Computational Cost |
|---|---|---|---|---|
| PBE-D3 | def2-TZVP | ~50 meV/atom [55] | High for vibrational frequencies [57] | Medium |
| B3LYP-D3 | def2-SVPD | ~50 meV/atom [55] | Reliable for pesticide identification [57] | Medium-High |
| B3LYP (uncorrected) | 6-31G* | Several hundred meV/atom [23] [55] | Poor for structural prediction [23] | Medium |
| LOSC | varies | Significant reduction in delocalization error [54] | Accurate quasiparticle energies [54] | Medium-High |
| r²SCAN-3c | def2-mTZVP | Improved over B3LYP/6-31G* [23] | Good for geometric structures [23] | Low-Medium |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of DFT correction methods requires careful selection of computational tools and experimental materials. The following table details essential components for establishing validated spectroscopic detection protocols.
Table 3: Essential Research Materials for DFT Spectral Validation
| Item/Category | Specific Examples | Function/Role in Workflow |
|---|---|---|
| Dispersion-Corrected Functionals | DFT-D3(BJ), DFT-D4 [56] | Account for van der Waals interactions critical for molecular recognition |
| Composite Methods | B3LYP-3c, r²SCAN-3c [23] | Provide balanced accuracy and efficiency for large systems |
| Vibrational Spectroscopy | Raman Spectroscopy, SERS [57] [28] | Experimental technique for acquiring reference spectra of contaminants |
| SERS Substrates | SiO₂ core-Au shell nanoparticles [5] | Enhance detection sensitivity for trace-level environmental contaminants |
| Machine Learning Algorithms | PCA, t-SNE [57] [28] | Classify spectral data and identify patterns in complex environmental samples |
| Solvent Systems | Acetone, toluene, hexane:acetone mixtures [5] | Extract contaminants from soil/water matrices with minimal spectral interference |
Diagram 2: DFT Error Correction Method Relationships
Best-Practice Recommendations for Environmental Applications
Based on comparative performance and validation studies, the following protocols represent current best practices for different scenarios in environmental contaminant research:
For High-Accuracy Spectral Prediction
Employ dispersion-corrected hybrid functionals (e.g., B3LYP-D3) with triple-zeta basis sets for predicting Raman spectra of environmental contaminants. This approach has demonstrated success in establishing reliable spectral databases for 166 pesticides and multiple PFAS compounds [57] [28]. The dispersion correction is essential for proper description of molecular interactions in complex environmental matrices.
For Large-Scale Screening
Utilize modern composite methods like r²SCAN-3c or B97M-V/def2-SVPD with built-in dispersion corrections for screening large databases of potential contaminants [23]. These methods provide an optimal balance between accuracy and computational efficiency, overcoming the limitations of outdated combinations like B3LYP/6-31G* that suffer from severe inherent errors including missing dispersion effects and basis set superposition error.
For Uncertainty Quantification
Implement emerging frameworks for quantifying uncertainty in DFT energy corrections, particularly when assessing phase stability or contaminant degradation pathways [55]. These methods account for both experimental uncertainty and parameter sensitivity, providing probability estimates for compound stability that enable better-informed assessments in environmental forensics.
The validation of DFT-calculated spectra for environmental contaminant detection depends critically on addressing systematic errors through carefully selected correction methods. Empirical dispersion corrections provide essential improvements for intermolecular interactions, while scaling corrections address fundamental delocalization error. Through rigorous experimental protocols incorporating machine learning validation and uncertainty quantification, researchers can establish reliable computational frameworks for detecting pesticides, PFAS, and other hazardous environmental contaminants. The continuing development of both empirical and first-principles corrections promises further enhancements in the accuracy and reliability of computational spectroscopy for environmental protection.
Density Functional Theory (DFT) is a cornerstone of computational materials science and chemistry. However, the accuracy of its predictions is fundamentally tied to the choice of the exchange-correlation (XC) functional. Standard functionals, like those within the Generalized Gradient Approximation (GGA), often fail to describe key phenomena such as van der Waals interactions and electronic properties of systems with localized d- or f-electrons. These limitations are particularly critical in environmental contaminant detection research, where accurately predicting interaction strengths and spectroscopic signatures is essential for developing reliable sensors.
This guide provides an objective comparison of two advanced strategies to overcome these limitations: the use of hybrid functionals, which incorporate a portion of exact Hartree-Fock exchange, and dispersion corrections, which explicitly account for long-range electron correlation effects. We will compare their performance against standard functionals and with each other, providing supporting data and detailed protocols to guide researchers in selecting the optimal method for their specific application in environmental sensing.
Theoretical Background and Key Concepts
Hybrid Functionals
Hybrid functionals mix the Hartree-Fock (HF) theory with DFT. A common form, such as in the popular B3LYP functional, combines a GGA functional with a set percentage of exact HF exchange. Range-separated hybrids (RSHs), like CAM-B3LYP, HSE06, and the ωB97 family, take this a step further by treating short- and long-range electron interactions differently, typically applying HF exchange more heavily at long range. This improves the description of electronic properties, most notably band gaps, which are systematically underestimated by GGA functionals [58] [59].
Dispersion Corrections
Dispersion interactions are weak, attractive forces arising from correlated electron movements between molecules. Standard DFT functionals fail to capture these effects. Dispersion corrections, such as the Grimme's D3 and D4 schemes, add an empirical, atom-pairwise correction term (e.g., -C₆R⁻⁶) to the total DFT energy. This is crucial for modeling the adsorption of contaminant molecules on sensor surfaces, as these interactions often dominate the binding process [60] [61] [62].
Performance Comparison of Computational Methods
Electronic Properties: Band Gap Accuracy
The accuracy of a material's band gap is vital for predicting the electronic response of chemiresistive sensors. Hybrid functionals offer a significant improvement over GGA.
Table 1: Mean Absolute Error (MAE) of Band Gap Predictions (eV)
| Material Class | GGA (PBE) | Hybrid (HSE06) | Reference |
|---|---|---|---|
| Binary Solids (121 materials) | 1.35 eV | 0.62 eV | Experimental data curated by Borlido et al. [58] |
A large-scale database of 7,024 inorganic materials demonstrated that the hybrid functional HSE06 corrects the band gap underestimation typical of GGA (here, PBEsol), shifting the values toward higher, more accurate ranges with a Mean Absolute Deviation (MAD) of 0.77 eV between the two methods [58] [59]. For 342 materials, PBEsol predicted metallic behavior while HSE06 correctly identified a finite band gap (≥ 0.5 eV) [58].
Geometric Parameters and Energetics
For structural properties and reaction energies, the combination of a standard functional with a dispersion correction often provides the best balance of accuracy and computational cost.
Table 2: Performance for Geometries and Energetics in Organometallics
| Functional Class | Example(s) | Performance for Metal-Carbonyl Bond Lengths | Performance for Relative Energies |
|---|---|---|---|
| GGA | BP86, PBE | Good with dispersion | Variable, can be poor |
| Hybrid | B3LYP | Good with dispersion | Good for thermochemistry |
| meta-GGA / Hybrid meta-GGA | r2SCAN, TPSSh | Best with dispersion (D3BJ/D4, D3zero) | Excellent, matches high-level DLPNO-CCSD(T) references [61] |
A benchmark study on Mn(I) and Re(I) carbonyl complexes found that meta-GGA and hybrid meta-GGA functionals, particularly r2SCAN(D3BJ/D4) and TPSSh(D3zero), provided the most reliable structures, vibrational properties, and energetics compared to high-level wavefunction theory [61]. The study evaluated 54 functional/dispersion combinations, highlighting the critical importance of including dispersion for non-covalent interactions.
Non-Covalent Interactions and Adsorption Strength
Dispersion corrections are indispensable for quantifying the adsorption of environmental contaminants on sensor materials.
Table 3: Adsorption Energies of Contaminants on Sensor Materials
| Adsorbent | Target Contaminant | Functional | Adsorption Energy (eV) | Key Interaction Types |
|---|---|---|---|---|
| MBTS Molecule [62] | Organophosphates (e.g., Malathion) | PBE-D3BJ | 0.27 - 1.05 eV | Hydrogen bonding, chalcogen bonding |
| Cu-Paddlewheel (MOF) [60] | Organic Solvent Vapors (e.g., THF) | B3LYP | ~ -1.12 eV (≈ -25.7 kcal/mol) | Coordination to open metal site, dispersion |
| Zn-doped C₆₀ [63] | Acetone | B97D | -0.47 eV (Strong, reversible) | Charge transfer, non-covalent |
Studies on organophosphate adsorption on modified graphene surfaces consistently use dispersion-corrected functionals (e.g., PBE-D3BJ) to capture the interplay of π-π stacking, hydrogen bonding, and van der Waals forces [62]. The omission of dispersion corrections leads to a severe overestimation of equilibrium distances and a complete lack of binding in physisorbed systems.
Magnetic Properties
The performance of functionals for calculating magnetic exchange coupling constants (J) in transition metal complexes is nuanced. A study on di-nuclear Cu and V complexes found that Scuseria-type range-separated functionals (e.g., HSE), which have a moderately low fraction of short-range HF exchange and no long-range HF exchange, outperformed the standard B3LYP functional in predicting J values [64]. This indicates that a very high fraction of HF exchange can be detrimental for accurately modeling these magnetic properties.
Experimental Protocols for Method Validation
Protocol: Validating DFT-Calculated Raman Spectra for Contaminant Detection
This protocol, derived from a study on polycyclic aromatic hydrocarbons (PAHs) in soil [36] [5], outlines how to validate DFT-calculated spectra against experimental data for contaminant identification.
Computational Spectral Prediction:
- Software: Use quantum chemistry packages (e.g., ORCA, Gaussian).
- Method Selection: Employ a hybrid functional (e.g., B3LYP, ωB97X) with a dispersion correction (e.g., D3BJ) and a polarized basis set (e.g., def2-SVP).
- Calculation: Perform geometry optimization and frequency calculation on the target contaminant molecule to obtain its theoretical Raman spectrum.
Experimental Data Acquisition:
- Technique: Use Surface-Enhanced Raman Spectroscopy (SERS) on nanostructured substrates (e.g., Au/SiO₂ nanoshells) to enhance signal.
- Sample Prep: Extract contaminants (e.g., PAHs) from soil matrices using solvent extraction (e.g., acetone via filtration or accelerated solvent extraction).
- Measurement: Deposit extract on SERS substrate and collect multiple spectra.
Machine Learning-Enabled Validation:
- Feature Extraction: Process both theoretical and experimental spectra using the Characteristic Peak Extraction (CaPE) algorithm to isolate distinctive spectral features, mitigating background interference and substrate-induced shifts.
- Similarity Analysis: Use the Characteristic Peak Similarity (CaPSim) algorithm to quantitatively compare the CaPE-processed theoretical and experimental spectra. A high similarity value (>0.6) validates the DFT protocol [5].
Protocol: Benchmarking DFT Methods for Organometallic Sensors
This protocol is based on benchmarking studies for metal-organic frameworks (MOFs) and metal carbonyl complexes [60] [61].
- System Selection: Choose a well-characterized model system, such as the copper paddlewheel node of a MOF or a fac-M(CO)₃L₃ complex (M = Mn, Re).
- Geometry Optimization:
- Test multiple functionals (e.g., GGA: PBE; Hybrid: B3LYP, HSE06; meta-GGA: r2SCAN) with and without dispersion corrections (D3BJ, D4).
- Use a core-level basis set (e.g., LANL2DZ for metals) and a polarized basis set (e.g., 6-31G*) for light atoms.
- Property Calculation:
- Adsorption Energy: For a contaminant molecule (e.g., THF, acetone), calculate Eads = Esystem - (Esensor + Econtaminant). Apply counterpoise correction for Basis Set Superposition Error (BSSE) [62].
- Electronic Properties: Calculate the HOMO-LUMO gap and density of states (DOS) for the sensor and sensor-contaminant complex.
- Validation against Reference:
- Structural: Compare optimized bond lengths (e.g., M-C, C≡O) and angles against high-quality crystallographic data from the Cambridge Structural Database (CCDC).
- Spectroscopic: Compare calculated vibrational frequencies (e.g., C≡O stretches) with experimental infrared or Raman spectra.
- Energetic: Compare relative energies with results from high-level ab initio methods like DLPNO-CCSD(T) [61].
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 4: Key Computational and Experimental Resources for Sensor Development
| Item Name | Function/Description | Example Use Case in Contaminant Detection |
|---|---|---|
| HSE06 Functional | A range-separated hybrid functional. Provides accurate electronic properties like band gaps for solids and surfaces. | Calculating the band structure of metal oxide sensors for improved accuracy over GGA [58] [59]. |
| D3/D4 Dispersion Corrections | Empirical corrections (Grimme) added to DFT energy to account for van der Waals forces. | Modeling the physisorption of organic contaminants (e.g., PAHs, solvents) on graphene or MOF surfaces [60] [61] [62]. |
| B3LYP-D3BJ Functional | A global hybrid functional combined with Becke-Johnson damping for dispersion. A versatile choice for molecular systems. | Studying the adsorption of organophosphate pesticides on functionalized graphene [62]. |
| Au/SiO₂ Nanoshells | Core-shell nanoparticles used as substrates for Surface-Enhanced Raman Spectroscopy (SERS). | Amplifying the Raman signal of trace-level PAHs in contaminated soil extracts for detection and validation [5]. |
| def2-SVP / def2-TZVP Basis Sets | Polarized Gaussian-type basis sets offering a good balance of accuracy and computational cost for molecular systems. | Geometry optimization and frequency calculations for contaminant molecules and their complexes with sensor materials [62]. |
| CPCM/SMD Solvation Models | Implicit solvation models to simulate the effect of a solvent (e.g., water) on the molecular system. | Modeling the adsorption of pollutants in aqueous environments, crucial for realistic sensor simulations [62] [63]. |
The strategic selection of DFT methodologies is paramount for the accurate prediction of material properties and molecular interactions in environmental sensor development. The evidence presented in this guide leads to the following conclusions:
- For Electronic Properties and Band Gaps: Hybrid functionals, particularly range-separated hybrids like HSE06, are unequivocally superior to GGA functionals. They correct the systematic underestimation of band gaps, a critical parameter for electronic sensors, with error reductions of over 50% compared to experiment [58].
- For Non-Covalent Interactions and Adsorption: The inclusion of empirical dispersion corrections (e.g., D3, D4) is non-negotiable. They are essential for quantitatively describing the adsorption of environmental contaminants on sensor surfaces, which is often governed by van der Waals forces [60] [61] [62].
- For a Balanced and Accurate Approach: No single functional is best for all properties. However, modern meta-GGAs and hybrid meta-GGAs like r2SCAN and TPSSh, when combined with an appropriate dispersion correction, offer an excellent compromise, delivering high accuracy for geometries, energies, and vibrational frequencies at a reasonable computational cost [61].
Therefore, the "advanced strategy" is not merely to use these tools, but to select them judiciously based on the target property—opting for hybrids for electronic structure and dispersion-corrected functionals for interaction energies—and to always validate the computational protocol against robust experimental or high-level theoretical benchmarks. This rigorous approach ensures reliable predictions that can accelerate the design of effective sensors for environmental monitoring.
Benchmarking and Validation: Ensuring Reliability Against Experimental Data
The validation of density functional theory (DFT) calculations against experimental data represents a critical step in developing reliable spectroscopic methods for environmental monitoring. For persistent pollutants like per- and polyfluoroalkyl substances (PFAS) and polycyclic aromatic hydrocarbons (PAHs), the ability to accurately predict vibrational and electronic spectra computationally enables more efficient identification and monitoring strategies [3] [5]. This guide provides a comprehensive comparison of methodologies and metrics for evaluating the agreement between calculated and experimental spectral peaks, focusing specifically on applications in environmental contaminant detection.
Quantitative Comparison Metrics
Core Performance Metrics
Table 1: Key Metrics for Experimental-Computational Spectral Comparison
| Metric | Calculation Method | Optimal Range | Application Context |
|---|---|---|---|
| Root Mean Square Deviation (RMSD) | (\sqrt{\frac{\sum{i=1}^{n}(x{calc,i} - x_{exp,i})^2}{n}}) | Lower values indicate better agreement; Study reported 3.4–8.6 cm⁻¹ for PFAS [65] | Vibrational frequency validation (IR/Raman) |
| Spectral Similarity Value | Algorithm-specific (e.g., CaPSim >0.6 indicates strong similarity [5]) | >0.6 (strong similarity) | Pattern recognition for contaminant identification |
| Peak Position Deviation | (\Delta \omega = \omega{calc} - \omega{exp}) | Varies by system; Typically <10 cm⁻¹ for DFT with appropriate basis set [3] | Individual peak assignment validation |
| Area Ratio Precision | (RA = \frac{A1}{A2}) | (\sqrt{2}) × more precise than intensity ratios [66] | Concentration quantification in complex mixtures |
The precision of area ratios (RA) has been theoretically and experimentally demonstrated to surpass that of intensity ratios (RI) by a factor of (\sqrt{2}), making area-based metrics particularly valuable for quantitative analysis of environmental contaminants [66]. This enhanced precision stems from negative covariance between intensity and bandwidth parameters, which reduces overall variance in area measurements.
Performance in Environmental Contaminant Detection
Table 2: DFT Performance in Environmental Contaminant Spectral Prediction
| Contaminant Class | Representative Compounds | Reported RMSD | Computational Level | Application Reference |
|---|---|---|---|---|
| PFAS | PFBA, PFHpA, PFOA, PFNA, PFDA, PFDoA | 3.4–8.6 cm⁻¹ [65] | DFT with 6-311++G(d,p) basis set [3] | Environmental monitoring [28] |
| PAHs | Pyrene, Anthracene | Spectral similarity >0.6 [5] | DFT with 6-311++G(d,p) basis set [5] | Soil contamination detection |
| Heterocyclic Compounds | Pyridine-2,6-dicarboxylic acid | Good agreement (specific values not reported) [67] | B3LYP/6-311++G(d,p) [67] | Drug development precursors |
Experimental Protocols for Method Validation
Sample Preparation and Spectral Acquisition
For PFAS compounds, researchers have developed standardized protocols for acquiring high-quality Raman spectra. Samples are placed on stainless steel squares approximately 2 inches per side, and spectra are collected using a Raman spectrometer equipped with a 785 nm laser source, 1200 grooves/mm grating, and 50× objective lens [3]. The laser power is maintained at 100 mW with 10-second exposure time and 5 accumulations to ensure sufficient signal-to-noise ratio while preventing sample degradation [3].
For PAH detection in soil samples, contamination procedures involve spiking soil samples with controlled concentrations of target analytes (e.g., pyrene, anthracene) in acetone solvent, followed by sealing, shaking for approximately 2 minutes to enhance absorption, and drying at room temperature until complete solvent evaporation [5]. Extraction employs either accelerated solvent extraction (ASE) or simple filtration methods, with studies showing comparable performance between these techniques [5].
Reference Spectral Databases
The creation of standardized reference spectral databases for bulk compounds addresses a significant challenge in environmental detection. Prior to these efforts, the lack of reference spectra complicated peak assignment and vibrational mode identification, particularly in surface-enhanced Raman spectroscopy (SERS) studies where signal enhancement and spectral variability depend heavily on substrate design and surface interactions [3]. Auto-generated databases using tools like ChemDataExtractor have demonstrated promise for creating scalable spectral libraries, having extracted 18,309 records of experimentally determined UV/vis absorption maxima from 402,034 scientific documents [68].
Computational Methodologies
DFT Calculation Parameters
For PFAS compounds, DFT calculations successfully predicted vibrational modes and enabled precise assignments of experimental Raman peaks [3] [65]. Systematic Raman shifts linked to PFAS chain length and functional groups facilitated structural identification, with the integration of machine learning techniques providing enhanced classification capabilities [3].
In the study of pyridine-2,6-dicarboxylic acid, computational investigations employed DFT with the B3LYP functional and 6-311++G(d,p) basis set, demonstrating good agreement with experimental IR and Raman spectra [67]. The optimized molecular structure served as the foundation for subsequent calculations of vibrational frequencies, natural bond orbital (NBO) analysis, and molecular electrostatic potential (MEP) surface mapping [67].
Workflow Integration
The following diagram illustrates the integrated computational-experimental workflow for spectral validation:
Advanced Analysis Techniques
Machine Learning Integration
Unsupervised machine learning algorithms, including principal component analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE), have demonstrated significant utility in clustering and separating Raman spectra of PFAS compounds [3] [28]. These techniques reveal both structural similarities and unique functional group influences, enabling differentiation of compounds with subtle spectral differences [65]. For PAH detection, physics-informed machine learning pipelines employ characteristic peak extraction (CaPE) algorithms to isolate distinctive spectral features, followed by characteristic peak similarity (CaPSim) algorithms to identify analytes with high robustness to spectral shifts and amplitude variations [5].
Spectral Comparison Algorithms
Multiple algorithms are available for comparing experimental and computational spectra:
- Linear Unmixing (LU): Effective for extracting quantitative signal traces in hyperspectral imaging applications [69]
- Matched Filter (MF): Provides similar performance to linear unmixing for quantitative analysis [69]
- Spectral Angle Mapper (SAM): Measures spectral similarity based on angular metrics
- Constrained Energy Minimization (CEM): Useful for detecting specific signatures in complex mixtures
Comparative studies indicate that LU and MF algorithms provide similar linear responses to increasing analyte concentrations and can both be effectively used for excitation-scanning hyperspectral imaging [69].
Research Reagent Solutions
Table 3: Essential Research Materials for Spectral Validation Studies
| Reagent/Material | Specifications | Application Function |
|---|---|---|
| PFAS Compounds | PFHpA, PFOA, PFDA, PFNA, 3:3FTCA, PFDoA, NEtFOSE, PFHxS, PFBA [3] | Target analytes for method development |
| SERS Substrates | SiO₂ core-Au shell nanoparticles (165±17 nm) [5] | Signal enhancement for trace detection |
| DFT Software | B3LYP/6-311++G(d,p) level theory [67] | Theoretical spectrum generation |
| Reference Compounds | Pyridine-2,6-dicarboxylic acid [67] | Method validation and calibration |
Best Practices and Recommendations
Experimental Design Considerations
Proper spectral comparison requires strict control of variables to ensure chemically legitimate conclusions. Key factors include:
- Instrumental Parameters: Maintain consistent resolution, scan number, and apodization functions across measurements, as these significantly impact spectral appearance [70]
- Sample Preparation: Use identical techniques for sample and reference materials, as different methods (e.g., ATR vs. transmission) produce spectral variations [70]
- Signal Processing: Prioritize area ratios over intensity ratios for quantitative analysis when precision is critical [66]
Method Selection Guidelines
The following diagram outlines the decision process for selecting appropriate comparison metrics:
The integration of computational and experimental approaches provides a powerful framework for environmental contaminant detection. Validation using quantitative metrics such as RMSD, spectral similarity values, and area ratio precision establishes the reliability of DFT-calculated spectra for identifying PFAS, PAHs, and related environmental pollutants. As spectral databases expand and machine learning algorithms become more sophisticated, these validated computational approaches will play an increasingly vital role in environmental monitoring and public health protection.
The accurate detection and identification of environmental contaminants, such as polycyclic aromatic hydrocarbons (PAHs) and per-fluoroalkyl substances (PFAS), is a critical challenge in environmental health research. In this context, spectral databases have become indispensable tools for researchers, providing curated reference data to compare against experimental results. The validation of density functional theory (DFT)-calculated spectra represents a burgeoning area of research, bridging computational predictions with empirical observation. This guide objectively compares the capabilities of the U.S. Environmental Protection Agency's (EPA) Analytical Methods and Open Spectral (AMOS) database against other emerging approaches that leverage computationally generated spectral libraries, providing experimental data and methodologies to inform researcher selection for environmental contaminant detection.
The landscape of spectral resources for environmental analysis ranges from established regulatory databases to innovative research-oriented approaches. The table below summarizes the core characteristics of these complementary resources.
Table 1: Comparison of Spectral Data Resources for Environmental Contaminant Analysis
| Resource | Primary Function | Data Types | Key Strengths | Notable Limitations |
|---|---|---|---|---|
| EPA AMOS Database | Regulatory method repository & spectral data access | Mass spectrometry, NMR, IR spectra; Regulatory method documents (PDF) | Official EPA regulatory methods; Integration with DSSTox substance database; Direct links to original sources [71] | Limited DFT-calculated spectra; Focus on established analytical methods |
| DFT-Calculated Spectral Libraries (Research) | In silico reference library creation | DFT-calculated Raman/SERS spectra | Covers compounds lacking experimental standards; Overcomes synthesis challenges for rare/modified contaminants [5] [72] | Requires experimental validation; Computational resource demands |
| Hybrid DFT/ML Workflows | Machine learning-enhanced contaminant detection | Surface-Enhanced Raman Spectroscopy (SERS) with DFT-calculated references | Identifies PAHs in complex soil matrices; High discriminative capability for isomers [5] [72] | Pipeline complexity; Specialized expertise required |
Experimental Validation of DFT-Calculated Spectra
Core Validation Methodologies
The credibility of DFT-calculated spectra for environmental application hinges on robust experimental validation. Two prominent research approaches demonstrate this process:
Physics-Informed Machine Learning for PAH Detection: Researchers developed a two-stage pipeline to detect PAHs in contaminated soil. First, the Characteristic Peak Extraction (CaPE) algorithm isolates distinctive spectral features from experimental Surface-Enhanced Raman Spectroscopy (SERS) data. Subsequently, the Characteristic Peak Similarity (CaPSim) algorithm identifies analytes by comparing these features against a DFT-calculated Raman spectral library. This method demonstrated strong similarity values (>0.6) between DFT-calculated and experimental SERS spectra for multiple PAHs, confirming its discriminative capability in complex soil matrices [5].
Chemometric Analysis for PFAS Identification: Researchers computed and analyzed the Raman spectra of 40 significant PFAS compounds using DFT. They identified specific spectral regions linked to critical chemical bonds (C-C, CF₂, CF₃) and key functional groups (-COOH, -SO₃H, -SO₂NH₂). By applying Principal Component Analysis (PCA) to the DFT-calculated spectral data, they effectively distinguished between PFAS isomers, noting that longer carbon chains increased the number of observable Raman peaks, providing more data points for analysis [72].
Quantitative Performance Comparison
The table below summarizes experimental performance metrics reported for these DFT-validation approaches.
Table 2: Experimental Performance Metrics of DFT-Based Detection Methods
| Method | Target Contaminants | Sample Matrix | Key Performance Metrics | Reference |
|---|---|---|---|---|
| CaPE/CaPSim with DFT | Pyrene, Anthracene | Soil extracts (43% clay, 37% sand) | Similarity values >0.6 vs. experimental SERS; Detection in complex soil background [5] | PNAS (2025) |
| DFT with Chemometrics | 40 PFAS compounds (PFOA, PFOS isomers) | Standard solutions | Identification of isomer-specific peak shifts in 200-800 cm⁻¹ and 1000-1400 cm⁻¹ regions [72] | Journal of Hazardous Materials (2024) |
| Δ-DFT Machine Learning | General molecular systems | Gas-phase simulations | Quantum chemical accuracy (<1 kcal·mol⁻¹ error); Corrected DFT-based MD simulations [73] | Nature Communications (2020) |
Research Workflow: Validating DFT-Calculated Spectra
The following diagram illustrates the conceptual workflow for validating DFT-calculated spectra against experimental data, integrating database resources and computational approaches.
Diagram 1: DFT Spectrum Validation Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of spectral validation requires specific materials and computational tools. The table below details essential components for these research workflows.
Table 3: Essential Research Reagents and Materials for Spectral Validation Studies
| Category | Specific Items | Function/Purpose | Example Applications |
|---|---|---|---|
| SERS Substrates | SiO₂ core-Au shell nanoparticles (nanoshells) | Signal enhancement for trace detection; 6-9 orders of magnitude signal improvement [72] | PAH detection in soil extracts; Trace PFAS analysis [5] [72] |
| Extraction Solvents | Acetone, toluene, 1:1 hexane:acetone, dichloromethane (DCM) | Contaminant isolation from environmental matrices; Acetone preferred for simpler Raman background [5] | Soil PAH extraction (filtration or accelerated solvent extraction) [5] |
| Computational Methods | Density Functional Theory (DFT); TD-DFT/CAM-B3LYP/6-31+G(d) | In silico spectral generation; Solvation effects modeling (IEFPCM) [74] | Prediction of UV/Vis absorption; Raman spectrum calculation [74] [72] |
| Machine Learning Algorithms | Characteristic Peak Extraction (CaPE); Characteristic Peak Similarity (CaPSim); Δ-DFT | Spectral feature isolation; DFT error correction; Quantum chemical accuracy attainment [5] [73] | PAH identification in complex matrices; CCSD(T)-accurate energies from DFT [5] [73] |
| Reference Databases | EPA AMOS; DSSTox Substance Database | Regulatory method context; Substance identifier mapping (DTXSID, CASRN) [71] | Method verification; Compound identification confirmation [71] |
The EPA AMOS database provides an essential foundation of regulatory methods and experimentally derived spectral data, particularly for mass spectrometry applications [71]. Meanwhile, emerging research demonstrates that DFT-calculated spectra, when validated through robust experimental workflows and machine learning algorithms, offer powerful capabilities for detecting environmental contaminants that challenge traditional methods [5] [72]. The most effective approach for environmental contaminant detection research often involves strategic integration of both resources: leveraging the verified experimental data in AMOS while supplementing with in silico spectral libraries for compounds lacking commercial standards. As machine learning methodologies continue to advance, particularly Δ-learning techniques that efficiently correct DFT errors [73], the integration of computational and experimental spectral data promises to significantly enhance environmental monitoring and public health protection.
The accurate identification of environmental contaminants, from persistent per- and polyfluoroalkyl substances (PFAS) to polycyclic aromatic hydrocarbons (PAHs), represents a critical challenge in modern analytical science. Traditional detection methods often struggle with the requirements for speed, sensitivity, and the ability to identify previously uncharacterized compounds. The integration of Density Functional Theory (DFT) and Machine Learning (ML) has emerged as a transformative approach, creating robust computational frameworks that enhance and accelerate the detection of hazardous substances. This synergy leverages the quantum-mechanical accuracy of DFT in predicting molecular properties with the pattern-recognition power of ML to interpret complex spectroscopic data, thereby validating detection results with unprecedented reliability. Within environmental contaminant research, this hybrid methodology is rapidly establishing a new standard for detection protocol validation, moving beyond traditional laboratory comparisons to computationally-driven verification. This guide examines the performance of this integrated approach against traditional alternatives, detailing the experimental protocols and computational infrastructure that enable its successful application.
Comparative Performance: DFT-ML vs. Traditional Methods
Quantitative comparisons reveal the significant advantages of combining DFT with machine learning over conventional detection methodologies. The following data, synthesized from recent studies, demonstrates this performance gap across several key metrics.
Table 1: Performance Comparison of PFAS Detection Methods
| Method Category | Specific Technique | Key Performance Metric | Reported Result | Limitations |
|---|---|---|---|---|
| Traditional Lab | Liquid Chromatography-Mass Spectrometry (LC-MS) | High sensitivity and specificity | Industry Standard | Expensive, lab-bound, complex sample prep [3] |
| Traditional Field | Fourier-Transform Infrared (FTIR) Spectroscopy | Practicality and accessibility | Useful for characteristic bands | Challenged by water interference, difficulty distinguishing similar PFAS [3] |
| DFT-ML Enhanced | SERS with DFT & ML (PFOS) | Limit of Detection (LOD) | 4.28 ppt (parts-per-trillion) | Requires model training and computational resources [3] |
| DFT-ML Enhanced | SERS with DFT & ML (PFOA) | Limit of Detection (LOD) | 1 ppt (parts-per-trillion) | Requires model training and computational resources [3] |
| DFT-ML Enhanced | Raman with DFT & ML (General PFAS) | Differentiation Capability | Successful clustering of 9 PFAS by structure using PCA/t-SNE | Some broad/weak peaks from sample prep [3] [28] |
The performance of the DFT-ML framework extends beyond sensitivity to encompass identification prowess. For instance, a study on nine PFAS compounds with varying chain lengths and functional groups demonstrated that the combination of experimental Raman spectra with DFT calculations and unsupervised ML (PCA and t-SNE) enabled clear clustering and separation, "revealing both structural similarities and unique functional group influences" [3]. This capability is vital for environmental forensics, where understanding the exact identity of a contaminant is as crucial as its mere presence.
Furthermore, the DFT-ML framework shows exceptional utility in scenarios where experimental reference data is scarce. A project from Rice University developed a method combining surface-enhanced Raman spectroscopy with a spectral reference library constructed entirely using DFT. This approach overcame a critical limitation in environmental monitoring: the lack of experimental data for many pollutants. The method successfully identified PAHs in soil and was validated by "strong similarity values (>0.6) between DFT-calculated and experimental surface-enhanced Raman spectra," even for lesser-known pollutant molecules [6]. This demonstrates the framework's power to expand the scope of detectable contaminants beyond the limits of existing physical libraries.
Experimental Protocols: Implementing the DFT-ML Workflow
The application of the DFT-ML framework for detection follows a structured workflow, integrating computational and experimental components. The diagram below outlines the core logical process for robust contaminant detection.
Protocol 1: DFT-Based Spectral Prediction and Library Construction
This protocol focuses on generating a theoretical spectral library, which is a cornerstone of the framework [6].
- A. Molecular Structure Definition: The target contaminant molecules are constructed computationally. For PFAS, this involves defining carbon chains of varying lengths (e.g., C4 for PFBA to C12 for PFDoA) and different functional groups (e.g., carboxylic acid vs. sulfonic acid) [3].
- B. DFT Calculation Setup: Electronic structure calculations are performed using software such as Vienna Ab Initio Simulation Package (VASP) [75]. Critical parameters include the selection of an exchange-correlation functional (e.g., ωB97M-V with def2-TZVPD basis set, as used in the OMol25 dataset [76]), convergence criteria for energy and forces, and settings for property-specific outputs like vibrational frequencies for Raman spectra.
- C. Spectral Generation: The results of the DFT calculations are processed to predict spectroscopic properties. For Raman spectra, this involves calculating the derivatives of the polarizability tensor to simulate the spectral fingerprint, including peak positions, intensities, and widths [3].
- D. Library Curation: The calculated spectra are stored in a database, forming a comprehensive theoretical library. This library is designed to include known contaminants and their potential derivatives, which may be commercially unavailable or challenging to synthesize for experimental reference [6].
Protocol 2: ML-Enhanced Spectral Matching and Validation
This protocol uses machine learning to bridge the gap between theoretical predictions and experimental observations.
- A. Data Acquisition & Preprocessing: Experimental spectra are collected from field or lab samples. The data is then preprocessed to minimize noise, correct baselines, and normalize intensities. This step is critical for ensuring the quality of input data for the ML model [3] [77].
- B. Feature Extraction: Unsupervised ML algorithms like Principal Component Analysis (PCA) or t-distributed Stochastic Neighbor Embedding (t-SNE) are employed to reduce the dimensionality of the spectral data. This process extracts the most characteristic features, facilitating the differentiation between contaminants based on structural features like chain length and functional groups [3].
- C. Model Training & Matching: A machine learning model is trained to recognize the relationship between the DFT-calculated spectra and their corresponding molecular structures. The model learns to account for experimental artifacts and spectral shifts. In the Rice University study, this was achieved using a Characteristic Peak Similarity (CaPSim) algorithm, which identifies analytes with high robustness to spectral variations [6]. The trained model is then used to match new, unknown experimental spectra against the DFT-generated library for identification.
The Scientist's Toolkit: Essential Research Reagents & Solutions
Successful implementation of the DFT-ML framework relies on a suite of computational and experimental tools. The following table details the key components and their functions.
Table 2: Essential Reagents and Solutions for DFT-ML Detection Research
| Tool Category | Specific Tool/Reagent | Function in the Workflow |
|---|---|---|
| Computational Software | Vienna Ab Initio Simulation Package (VASP) [75] | Performs quantum-mechanical DFT calculations to predict electronic structure and molecular properties. |
| Computational Software | ORCA [76] | A quantum chemistry program used for high-precision DFT calculations, such as those generating the OMol25 dataset. |
| Computational Resource | High-Performance Computing (HPC) Cluster | Provides the computational power required for large-scale DFT calculations, which can consume billions of CPU core-hours [76]. |
| Reference Dataset | OMol25 Dataset [76] | Provides a large-scale, high-precision quantum chemistry dataset for training and benchmarking machine learning interatomic potentials. |
| ML Algorithm | Principal Component Analysis (PCA) / t-SNE [3] | Unsupervised learning methods for dimensionality reduction and clustering of spectral data to visualize and confirm differentiation. |
| ML Algorithm | Convolutional Neural Networks (CNNs) [77] | Deep learning models effective at classifying one-dimensional spectral data, robust to noise and background signals. |
| Experimental Substrate | Silver Nanoparticles (Ag NPs) / Nanostructured Surfaces | Used in Surface-Enhanced Raman Spectroscopy (SERS) to amplify the Raman signal of target molecules by several orders of magnitude [3]. |
| Target Analytes | PFAS Compounds (e.g., PFOA, PFOS, PFHxS) [3] | Model environmental contaminants used to develop and validate the DFT-ML detection framework. |
The integration of Density Functional Theory and Machine Learning represents a powerful and validated paradigm shift in detection science. As the comparative data and protocols outlined in this guide demonstrate, this hybrid framework does not merely supplement traditional methods but surpasses them in key areas: achieving ultra-trace detection limits, enabling the identification of compounds without existing experimental standards, and providing a robust, computationally-driven validation pathway. For researchers and drug development professionals, mastering this toolkit is no longer a niche specialty but an essential skill for tackling the next generation of challenges in environmental monitoring, forensics, and public health protection. The continued growth of high-quality computational datasets and more efficient algorithms promises to further solidify this approach as the gold standard for robust contaminant detection.
Density Functional Theory (DFT) stands as a cornerstone computational method in chemistry, physics, and materials science for investigating electronic structure. Its versatility allows for the study of diverse systems, from drug molecules to new materials [78]. Within environmental research, accurately identifying pollutants like polycyclic aromatic hydrocarbons (PAHs) in complex matrices such as soil is crucial for assessing public health risks. The validation of computational methods, particularly the use of DFT-calculated spectra for detecting these environmental contaminants, is therefore a pressing research topic [5]. This guide provides an objective comparison of DFT against other computational methodologies, focusing on performance metrics, computational complexity, and practical applications in environmental science. The analysis aims to equip researchers with the data needed to select the most appropriate tool for their specific challenges in contaminant detection and material design.
Performance Benchmarking: Accuracy and Speed
Accuracy Across Chemical Space
The accuracy of computational methods varies significantly across different chemical systems. Benchmark studies are essential for understanding their performance and limitations.
Table 1: Performance Comparison of Electronic Structure Methods for Transition Metal Systems
| Method Category | Representative Methods | Mean Unsigned Error (MUE) for Por21 Database (kcal/mol) | Performance Grade for Metalloporphyrins | Key Strengths | Key Limitations |
|---|---|---|---|---|---|
| Local DFT (GGA, meta-GGA) | GAM, r2SCAN, revM06-L [79] | <15.0 (Best performers) | A | Good for spin state energies; low computational cost [79] | Moderate accuracy for certain properties |
| Hybrid DFT (Low exact exchange) | r2SCANh, B98 [79] | ~15.0-23.0 | A-B | Improved accuracy over local functionals for some properties [79] | Higher cost than local functionals |
| Hybrid DFT (High exact exchange) | M06-2X, HFLYP [79] | >>23.0 | F | Can be good for main-group chemistry | Catastrophic failures for transition metal spin states [79] |
| Wavefunction Methods | CASPT2 [79] | Used as reference | N/A | High accuracy; treats multireference character | Extremely high computational cost; not for routine use [79] |
| Machine Learning-Enhanced DFT | Skala [80] | Reaches chemical accuracy (~1 kcal/mol) for main group molecules [80] | N/A | Reaches experimental accuracy; generalizes well [80] | Requires extensive training data; newer method |
For transition metal complexes like metalloporphyrins, a benchmark study of 250 electronic structure methods revealed that most approximations fail to achieve the "chemical accuracy" target of 1.0 kcal/mol. The best-performing DFT functionals achieved mean unsigned errors (MUEs) below 15.0 kcal/mol, but errors for most methods were at least twice as large [79]. Local functionals and global hybrids with a low percentage of exact exchange generally perform best for spin states and binding energies in these systems, whereas approximations with high exact exchange often lead to catastrophic failures [79].
In contrast, for main-group molecules, a breakthrough deep-learning approach has demonstrated the potential to overcome DFT's long-standing accuracy limitations. The novel Skala functional, trained on a large dataset of highly accurate wavefunction data, can reach the chemical accuracy required to reliably predict experimental outcomes for atomization energies, a fundamental thermochemical property [80].
Computational Efficiency and Scalability
Computational cost is a critical factor in method selection, especially for large systems or high-throughput screening.
Table 2: Computational Complexity and Efficiency Comparison
| Method Category | Computational Complexity | Key Efficiency Features | Practical Scaling |
|---|---|---|---|
| Traditional DFT | O(N³) [80] | Mature, widely implemented codes | Cubic scaling with system size |
| Accelerated DFT (GPU-cloud) | ~Order of magnitude speedup vs. CPU [78] | Cloud-native, API-driven; optimized for GPUs [78] | Efficient for small to medium molecules |
| Wavefunction Methods (e.g., CASPT2) | Exponential [80] | Necessary for multireference systems | Prohibitively expensive for large systems [79] |
| Discrete Fourier Transform (Signal Processing) | O(N²) [81] [82] | Efficient algorithms (FFT) available | Not directly comparable (different application domain) |
Traditional DFT calculations scale cubically with the number of electrons, a significant improvement over the exponential scaling of brute-force solutions to the many-electron Schrödinger equation [80]. Recent innovations leverage cloud infrastructure and GPU-first algorithm redesign to achieve an order-of-magnitude acceleration in DFT simulations compared to other programs using the same GPU or similar CPU cloud resources [78]. This cloud-native, service-based approach makes high-speed DFT calculations more accessible and scalable [78].
Experimental Protocols and Validation in Environmental Research
Workflow for Validating DFT-Calculated Spectra in Contaminant Detection
The following diagram illustrates the integrated physics-informed machine learning pipeline for detecting environmental contaminants using validated DFT-calculated spectra.
The experimental workflow for validating and applying DFT-calculated spectra in environmental detection involves a multi-stage process, as demonstrated in research on PAH detection in soil [5]:
- Sample Preparation and Contamination: Soil samples are artificially contaminated with specific PAHs (e.g., pyrene, anthracene) at controlled concentrations. The soil-PAH mixture is sealed, shaken to enhance absorption, and dried [5].
- Analyte Extraction: Contaminants are extracted from the soil using a solvent like acetone, which offers a simpler Raman signal background. Both simple filtration at room temperature and accelerated solvent extraction (ASE) have been shown to be effective, with the filtration method providing a more practical and accessible alternative [5].
- Spectral Data Acquisition: The extracted solution is deposited onto a Surface-enhanced Raman spectroscopy (SERS) substrate, such as SiO₂ core-Au shell nanoparticles (nanoshells), and drop-dried. Multiple SERS spectra are collected from different regions of the substrate to ensure reproducibility [5].
- DFT Spectral Calculation (In-silico Library): Theoretical Raman spectra for target contaminants are computed using DFT. This creates a ground-truth spectral library in silico, overcoming limitations of experimental libraries, such as spectral interference and the unavailability of certain compounds [5].
- Feature Extraction and Model Training: The Characteristic Peak Extraction (CaPE) algorithm processes both the experimental SERS spectra and the DFT-calculated spectra to isolate distinctive spectral features, providing tolerance to spectral shifts and amplitude variations. A machine learning model is then trained to differentiate between contaminated and reference soil samples using these extracted features [5].
This pipeline validates the DFT-calculated spectra against experimental SERS data and leverages them to accurately identify analytes in a complex environmental matrix.
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Materials for SERS-Based Environmental Detection with DFT Validation
| Item | Function/Description | Application Context |
|---|---|---|
| SERS Substrates | SiO₂ core-Au shell nanoparticles (nanoshells); provide plasmonic enhancement for Raman signal [5]. | Essential for acquiring high-sensitivity SERS spectra from trace analytes. |
| Reference Compounds | High-purity PAHs (e.g., pyrene, anthracene); used for controlled contamination and method validation [5]. | Creating ground-truthed experimental data. |
| Solvents for Extraction | Acetone, toluene, dichloromethane; used to extract contaminants from environmental matrices [5]. | Acetone is preferred for its simpler Raman background. |
| DFT Software | Accelerated DFT, various electronic structure codes; calculate theoretical Raman spectra [78] [5]. | Generating the in-silico spectral library for identification. |
| Feature Extraction Algorithms | Characteristic Peak Extraction (CaPE); isolates distinctive spectral features from complex data [5]. | Preprocessing step to improve robustness of machine learning models. |
The comparative analysis reveals that DFT holds a unique position in the computational toolkit. While it traditionally struggles with chemical accuracy for challenging systems like transition metals, its favorable scaling and computational efficiency make it vastly more practical than high-accuracy wavefunction methods for most applications. The emergence of AI-enhanced functionals like Skala signals a paradigm shift, potentially bridging the accuracy gap while retaining DFT's computational advantages [80]. In environmental research, the validation of DFT-calculated spectra has proven highly effective, enabling the creation of reliable in-silico libraries that are crucial for detecting harmful contaminants in complex samples like soil [5]. The integration of cloud-native, GPU-accelerated DFT platforms further promises to democratize access and speed up discoveries [78]. For researchers in environmental science and drug development, the choice of method must balance accuracy, cost, and system-specific requirements, with DFT—particularly in its modern, AI-driven incarnations—offering a powerful and increasingly predictive solution for a wide range of challenges.
Conclusion
The validation of DFT-calculated spectra represents a powerful and evolving paradigm for environmental contaminant detection. By understanding its foundational principles, meticulously applying and optimizing methodological workflows, and rigorously benchmarking results against experimental data, researchers can transform DFT from a theoretical tool into a reliable, predictive asset. Future directions point toward tighter integration with machine learning algorithms, the development of more specialized functionals for environmental applications, and the expansion of open-access spectral databases. These advances will further solidify DFT's role not only in environmental protection and remediation but also in the broader biomedical field for understanding pollutant interactions and aiding in the development of targeted therapeutics.
References
- 1. Image super-resolution inspired electron density prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC12102193/]
- 2. Comparison of the Performance of Density Functional ... [https://www.mdpi.com/1420-3049/28/8/3487]
- 3. Experimental Raman spectra analysis of selected PFAS ... [https://www.sciencedirect.com/science/article/abs/pii/S0304389425016206]
- 4. Experimental Raman spectra analysis of selected PFAS ... [https://pubmed.ncbi.nlm.nih.gov/40440902/]
- 5. In silico machine learning–enabled detection of polycyclic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12088428/]
- 6. Rice University spots soil contamination more rapidly [https://optics.org/news/16/5/18]
- 7. DFT analysis of structural, electronic and optical properties ... [https://www.nature.com/articles/s41598-025-19663-7]
- 8. Density Functional Theory Investigation on the Molecular Structure and Vibrational Spectra of Triclosan | Spectroscopy Online [https://www.spectroscopyonline.com/view/density-functional-theory-investigation-on-the-molecular-structure-and-vibrational-spectra-of-triclosan]
- 9. Extensive TDDFT Benchmark Study of the Resonance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495946/]
- 10. Reproducible Density Functional Theory Predictions of ... [https://www.sciencedirect.com/science/article/pii/S2352214325001224]
- 11. How Accurate Are DFT Forces? Unexpectedly Large ... [https://arxiv.org/html/2510.19774v1]
- 12. TD-DFT Guided Advanced E-Eye Sensing Technique for ... [https://pubmed.ncbi.nlm.nih.gov/41052021/]
- 13. and M-edge spectra using the DFT/CIS method with spin– ... [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp01656h]
- 14. Polycyclic Aromatic Hydrocarbons: Sources, Toxicity, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7674206/]
- 15. Unveiling the occurrence and ecological risks of triclosan ... [https://www.sciencedirect.com/science/article/pii/S0269749124016154]
- 16. Distribution and Health Risk Assessment of Triclosan and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12191833/]
- 17. Climate, Environmental, and Health Impacts of Fossil Fuels ... [https://www.eesi.org/papers/view/fact-sheet-climate-environmental-and-health-impacts-of-fossil-fuels-2021]
- 18. DFT-guided Raman spectral analysis and machine ... [https://pubmed.ncbi.nlm.nih.gov/41274641/]
- 19. Artificial intelligence technology in environmental research ... [https://www.sciencedirect.com/science/article/pii/S0160412025005392]
- 20. A perylene derivative for multi-hazard detection in ... [https://www.sciencedirect.com/science/article/pii/S2772416625002748]
- 21. DFT investigation of dye adsorption on pristine and doped ... [https://pubs.rsc.org/en/content/articlehtml/2025/na/d5na00720h]
- 22. Real-space Kohn–Sham density functional theory for complex ... [https://pubs.rsc.org/en/content/articlehtml/2025/cc/d5cc01820j?page=search]
- 23. Best‐Practice DFT Protocols for Basic Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9826355/]
- 24. DFT/TDDFT calculations of geometry optimization ... [https://www.nature.com/articles/s41598-024-58599-2]
- 25. Quantum chemistry composite methods [https://en.wikipedia.org/wiki/Quantum_chemistry_composite_methods]
- 26. Frequencies and Thermochemistry | Rowan Documentation [https://docs.rowansci.com/science/quantum-chemistry/frequencies-and-thermochemistry]
- 27. Let's benchmark quantum chemistry packages! [https://github.com/r2compchem/benchmark-qm]
- 28. Raman Spectroscopy and Machine Learning Show ... [https://www.spectroscopyonline.com/view/raman-spectroscopy-and-machine-learning-show-promise-for-pfas-detection]
- 29. An accurate and efficient framework for modelling the ... [https://www.nature.com/articles/s41557-025-01884-y]
- 30. Comparative analysis of the response surface ... [https://www.sciencedirect.com/science/article/pii/S2666086525000220]
- 31. Efficient removal of short- and long-chain perfluoroalkyl ... [https://pubs.rsc.org/en/content/articlehtml/2025/ew/d5ew00320b]
- 32. Improving the adsorption efficiency of a low-cost natural ... [https://pubs.rsc.org/en/content/articlehtml/2025/ma/d5ma00253b]
- 33. Experimental Studies of Adsorption Desalination Systems [https://www.sciencedirect.com/science/article/pii/S2590174525001485]
- 34. Protein Contaminants Matter: Building Universal Protein Contaminant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10040255/]
- 35. Simple statistical identification and removal of contaminant ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-018-0605-2]
- 36. Machine learning powers new approach to detecting soil ... [https://news.rice.edu/news/2025/machine-learning-powers-new-approach-detecting-soil-contaminants]
- 37. Predicting accurate fluorescent spectra for high molecular ... [https://www.sciencedirect.com/science/article/abs/pii/S0022285216301187]
- 38. In silico machine learning-enabled detection of polycyclic ... [https://pubmed.ncbi.nlm.nih.gov/40339117/]
- 39. Persistent polycyclic aromatic hydrocarbons (PAHs) in the soil ... [https://enveurope.springeropen.com/articles/10.1186/s12302-025-01230-6]
- 40. Fate of polycyclic aromatic hydrocarbons in the ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1550234/full]
- 41. Predicting thermal desorption efficiency of PAHs in ... [https://www.sciencedirect.com/science/article/abs/pii/S0269749124003816]
- 42. First principles prediction of two-dimensional metal-organic ... [https://archive.aps.org/mar/2021/b56/12/]
- 43. A comprehensive approach to incorporating intermolecular ... [https://www.sciencedirect.com/science/article/abs/pii/S0009250925009935]
- 44. Detecting charge transfer at defects in 2D materials with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12523977/]
- 45. Detecting charge transfer at defects in 2D materials with ... [https://arxiv.org/html/2301.04469v5]
- 46. function Density ( theory ) DFT infrared absorption calculated ... spectra [https://pubmed.ncbi.nlm.nih.gov/32144228/]
- 47. Development and validation of a multi-residue analytical ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967325008106]
- 48. Shock-induced dispersion patterns of powder with diverse ... [https://pubs.rsc.org/en/content/articlehtml/2025/sm/d4sm01541j]
- 49. Density Functional (DFT) Methods [https://gaussian.com/dft/]
- 50. Gaussian Basis Sets for Crystalline Solids: All-Purpose ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7997400/]
- 51. Basis Sets [https://gaussian.com/basissets/]
- 52. Using Cost-Effective Density Functional Theory (DFT) to ... [https://chemrxiv.org/engage/chemrxiv/article-details/60c7418d842e65304ddb1ee3]
- 53. Different Basis Sets for Gaussian Calculations [http://williamkennerly.com/blog/different-basis-sets-for-gaussian-calculations/]
- 54. Development and Application of Scaling Correction Methods ... [https://chem.duke.edu/events/development-and-application-scaling-correction-methods-density-functional-theory]
- 55. A framework for quantifying uncertainty in DFT energy ... [https://www.nature.com/articles/s41598-021-94550-5]
- 56. 5.7.3 Empirical Dispersion Corrections: DFT-D [https://manual.q-chem.com/6.0/subsec_DFT-D.html]
- 57. From theory to practice: DFT-guided Raman spectral ... [https://www.sciencedirect.com/science/article/pii/S209012322500921X]
- 58. Materials Database from All-electron Hybrid Functional ... [https://www.nature.com/articles/s41597-025-05867-z]
- 59. Materials Database from All-electron Hybrid Functional ... [https://arxiv.org/html/2504.20812v1]
- 60. Dispersion corrected-DFT investigation on the adsorption ... [https://www.sciencedirect.com/science/article/abs/pii/S0019452222004113]
- 61. Assessing Density Functionals and Dispersion Corrections for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12511928/]
- 62. DFT Insights into the Adsorption of Organophosphate ... [https://www.mdpi.com/2673-6918/5/4/43]
- 63. High-performance electrochemical sensors based on ... [https://www.nature.com/articles/s41598-025-24911-x]
- 64. The performance and efficiency of twelve range-separated ... [https://www.sciencedirect.com/science/article/pii/S2210271X2400080X]
- 65. New Publication: Raman Spectroscopy, DFT, and Machine ... [https://www.zhao-nano-lab.com/post/new-publication-raman-spectroscopy-dft-and-machine-learning-a-combined-approach-for-differentiati]
- 66. Precision comparison of intensity ratios and area ... [https://www.nature.com/articles/s41598-024-71653-3]
- 67. Experimental spectroscopic and quantum computational ... [https://www.sciencedirect.com/science/article/abs/pii/S0022286021011789]
- 68. Comparative dataset of experimental and computational ... [https://www.nature.com/articles/s41597-019-0306-0]
- 69. Comparing Performance of Spectral Image Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10295298/]
- 70. How to Properly Compare Spectra, and Determining ... [https://www.spectroscopyonline.com/view/how-properly-compare-spectra-and-determining-alkane-chain-length-infrared-spectra]
- 71. Analytical Methods and Open Spectral (AMOS) Database ... [https://www.epa.gov/comptox-tools/analytical-methods-and-open-spectral-amos-database-user-guide]
- 72. Breakthrough Raman Spectroscopy Study Reveals Key to ... [https://www.spectroscopyonline.com/view/breakthrough-raman-spectroscopy-study-reveals-key-to-advanced-pfas-detection]
- 73. Quantum chemical accuracy from density functional ... [https://www.nature.com/articles/s41467-020-19093-1]
- 74. Experimental and DFT/TD-DFT computational ... [https://www.sciencedirect.com/science/article/pii/S0167732220328889]
- 75. A deep learning framework to emulate density functional ... [https://www.nature.com/articles/s41524-023-01115-3]
- 76. OMol25: High-Precision Molecular DFT Dataset [https://www.emergentmind.com/topics/omol25-dataset]
- 77. Validating neural networks for spectroscopic classification ... [https://www.nature.com/articles/s41524-023-01055-y]
- 78. Acceleration without Disruption: DFT Software as a Service [https://arxiv.org/html/2406.11185v1]
- 79. Comparison of the Performance of Density Functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10146789/]
- 80. Breaking bonds, breaking ground: Advancing the accuracy ... [https://www.microsoft.com/en-us/research/blog/breaking-bonds-breaking-ground-advancing-the-accuracy-of-computational-chemistry-with-deep-learning/]
- 81. 5.8: DFT - Computational Complexity [https://eng.libretexts.org/Bookshelves/Electrical_Engineering/Introductory_Electrical_Engineering/Electrical_Engineering_(Johnson)/05%3A_Digital_Signal_Processing/5.08%3A_DFT_-_Computational_Complexity]
- 82. DFT: Computational Complexity [https://www.opentextbooks.org.hk/ditatopic/9757]